












网站维护
系统内容更新/升级中


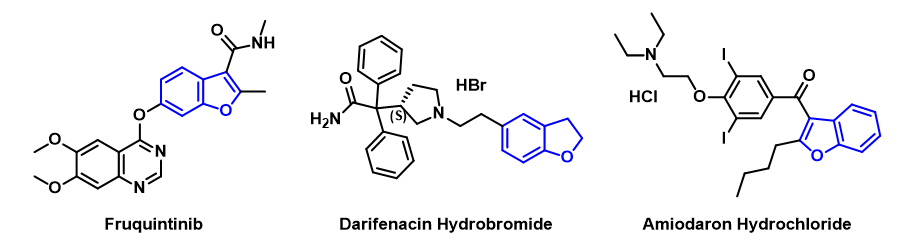
Benzofuran is an important class of heterocyclic compounds and a common motif in pharmaceuticals. Its synthesis has a wide range of applications in drug discoveries. For example, Amiodarone, an antiarrhythmic drug developed by Pfizer, Darifenacin hydrobromide, a genitourinary drug marketed by Novartis, and Fruquintinib, an antineoplastic drug developed by Hutchison Whampoa (Figure 1).[1]
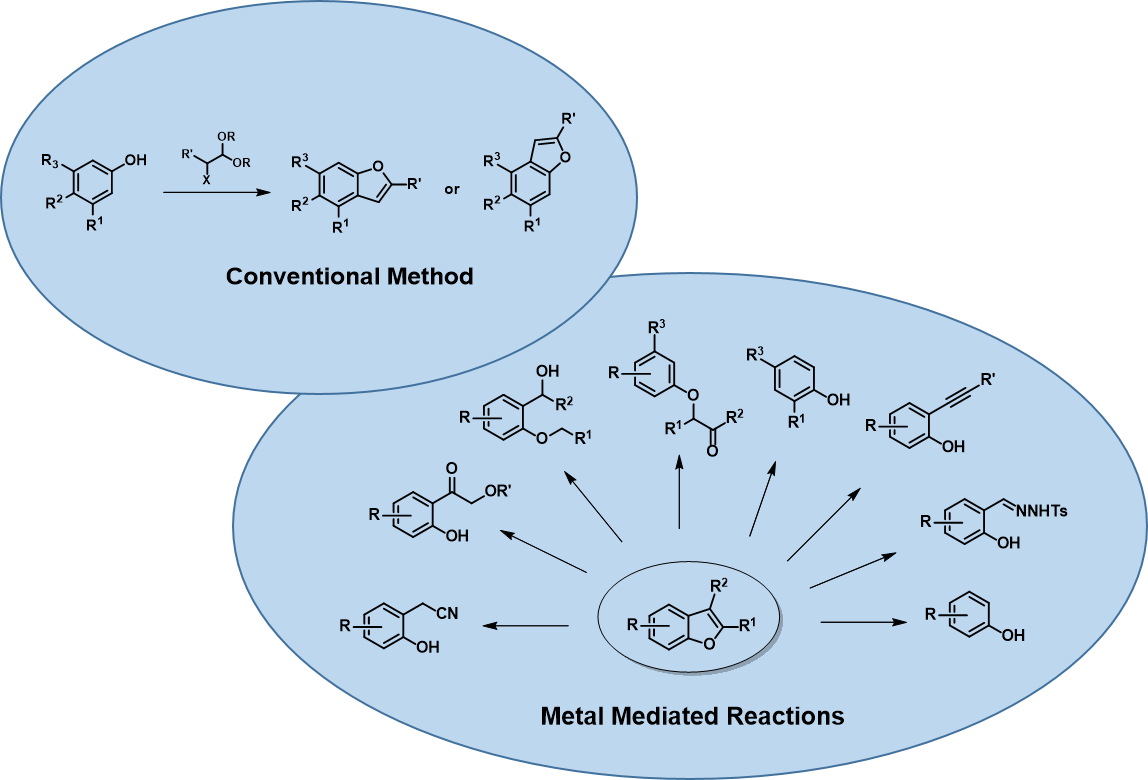
There are many ways to construct the benzofuran scaffold[2]; among them, cyclization of aryl ether substrates is the most popular synthetic method used in our laboratory (Figure 2). In this chapter, we’ll discuss the use of molecular orbital analysis to predict regioselectivity of acid-catalyzed cyclization of the acetal substrates.
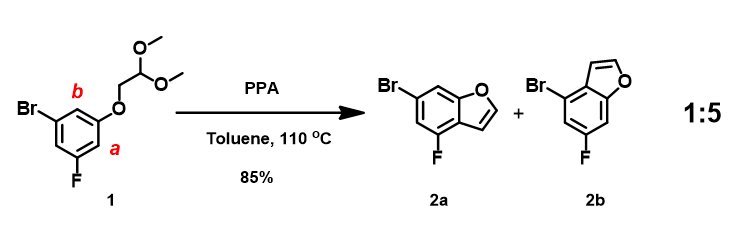
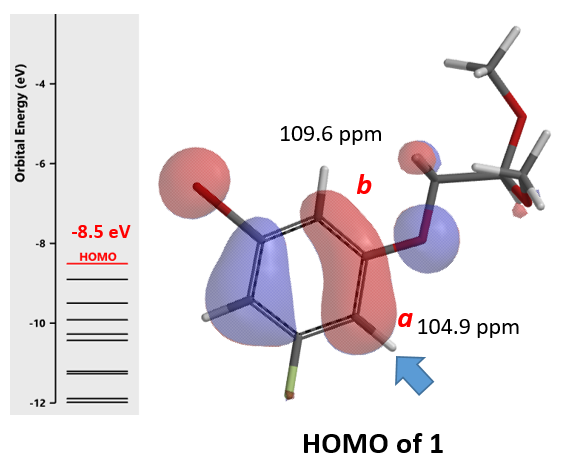
In our laboratory, we synthesized benzofuran core 2a (Figure 3) via polyphosphoric acid (PPA) catalyzed cyclization of acetal 1. QM analyses (HOMO and 13C NMR calculation) of 1 suggested that the reaction will provide a mixture of regioisomers 2a and 2b, with 2a being the major product. Experimentally, a 1:5 mixture is observed, with 2b as the major product (Figure 4), requiring us to reassess QM analysis of this type of reaction.
Mechanistically, substrate 1 is first protonated under acidic conditions, followed by elimination of MeOH to form oxonium ion A. The phenyl ring underwent nucleophilic addition, at sites a and b, to provide B and C, followed by a second MeOH elimination to provide a mixture of products, 2a and 2b (Figure 5).
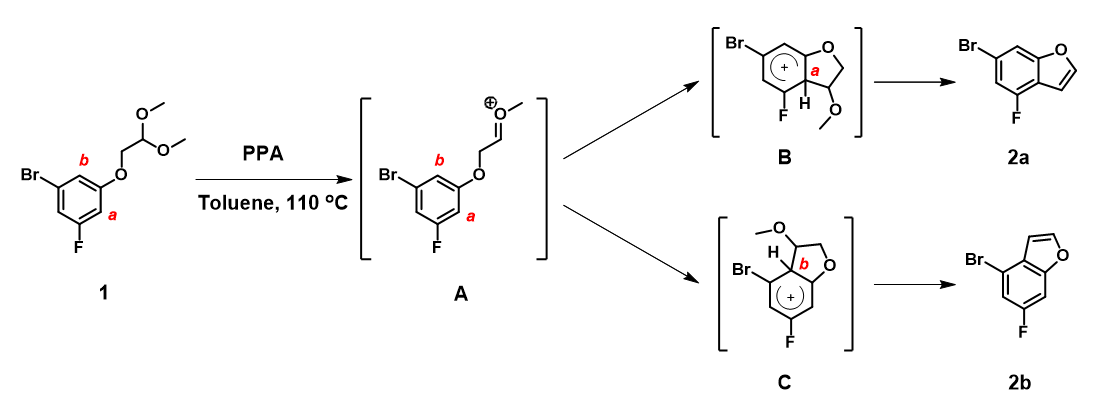
As such, it is more relevant to look at properties of oxonium ion A to assess regioselectivity of the cyclization.
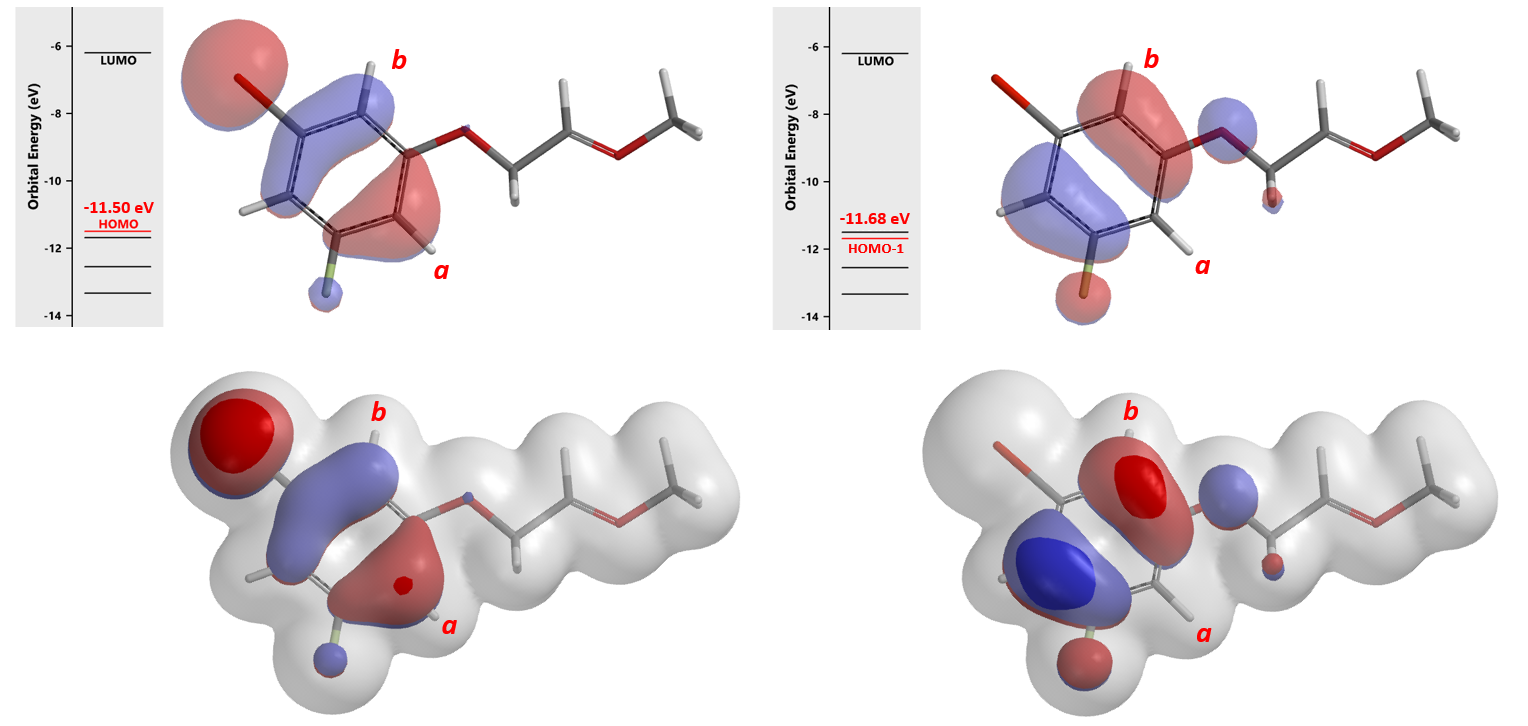
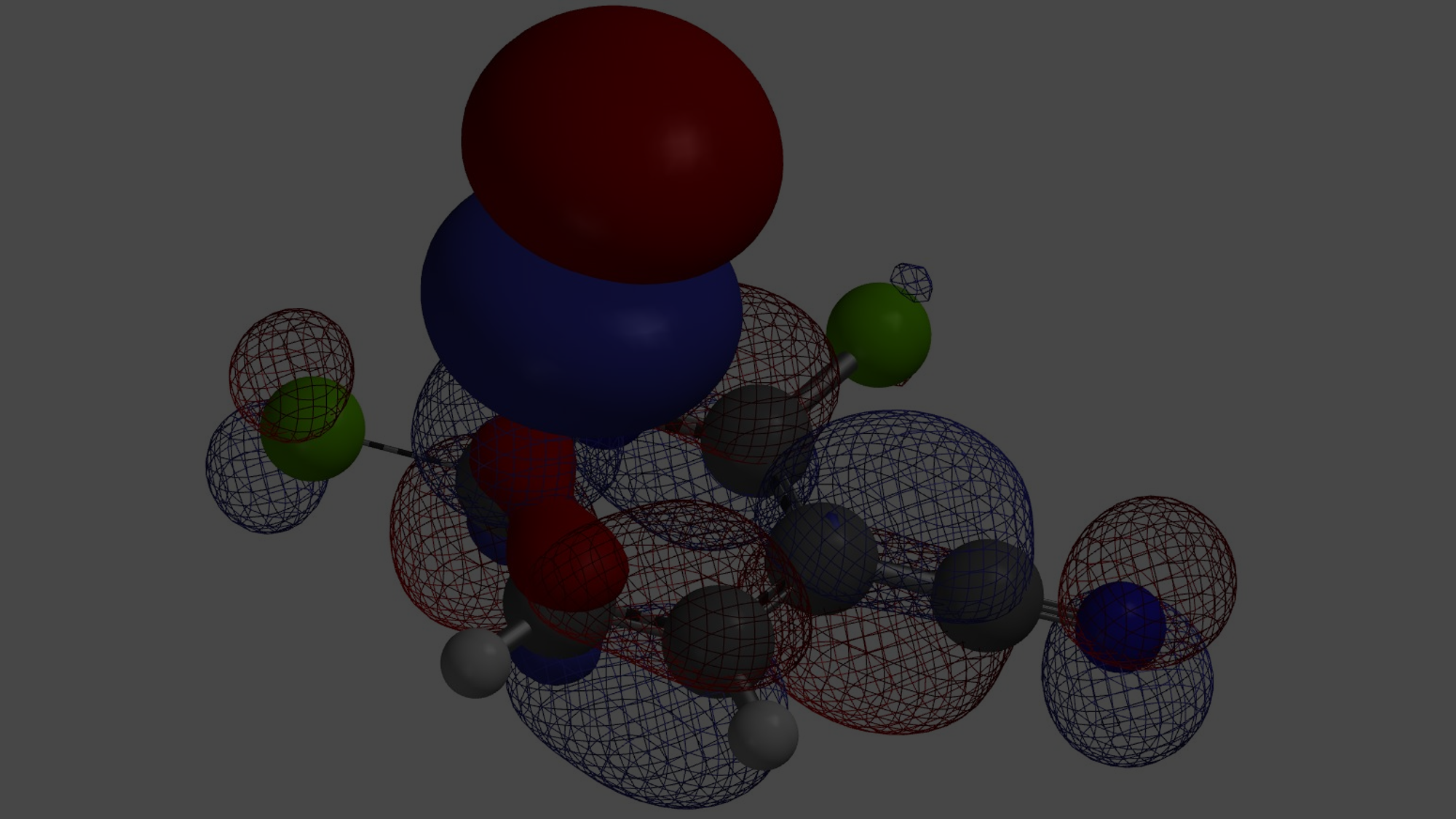
Shown in Figure 6 are the HOMO (-11.50 eV) and HOMO-1 (-11.68 eV) calculated for oxonium ion A. Since energy difference between the two orbitals is small, 0.18 eV, we need to consider both of them for subsequent cyclization. In this situation, we learned to superimpose HOMO and HOMO-1 onto Electron Density Surface (EDS)[3] of A for further evaluation. The electron density isosurface-orbital overlays revealed that HOMO-1 lobe at site b is a lot more accessible than HOMO lobe at site a, suggesting that 2b could be the major product.
Concurrently, we calculated for the reaction energy profiles for the two competition pathways. As shown in Figure 7, activation energy required for the cyclization reaction at site b is ~9.22 kcal/mol, which is 0.94 kcal/mol lower than the 10.16 kcal/mol required for reaction at site a, substantiating the orbital-EDS analyses above.
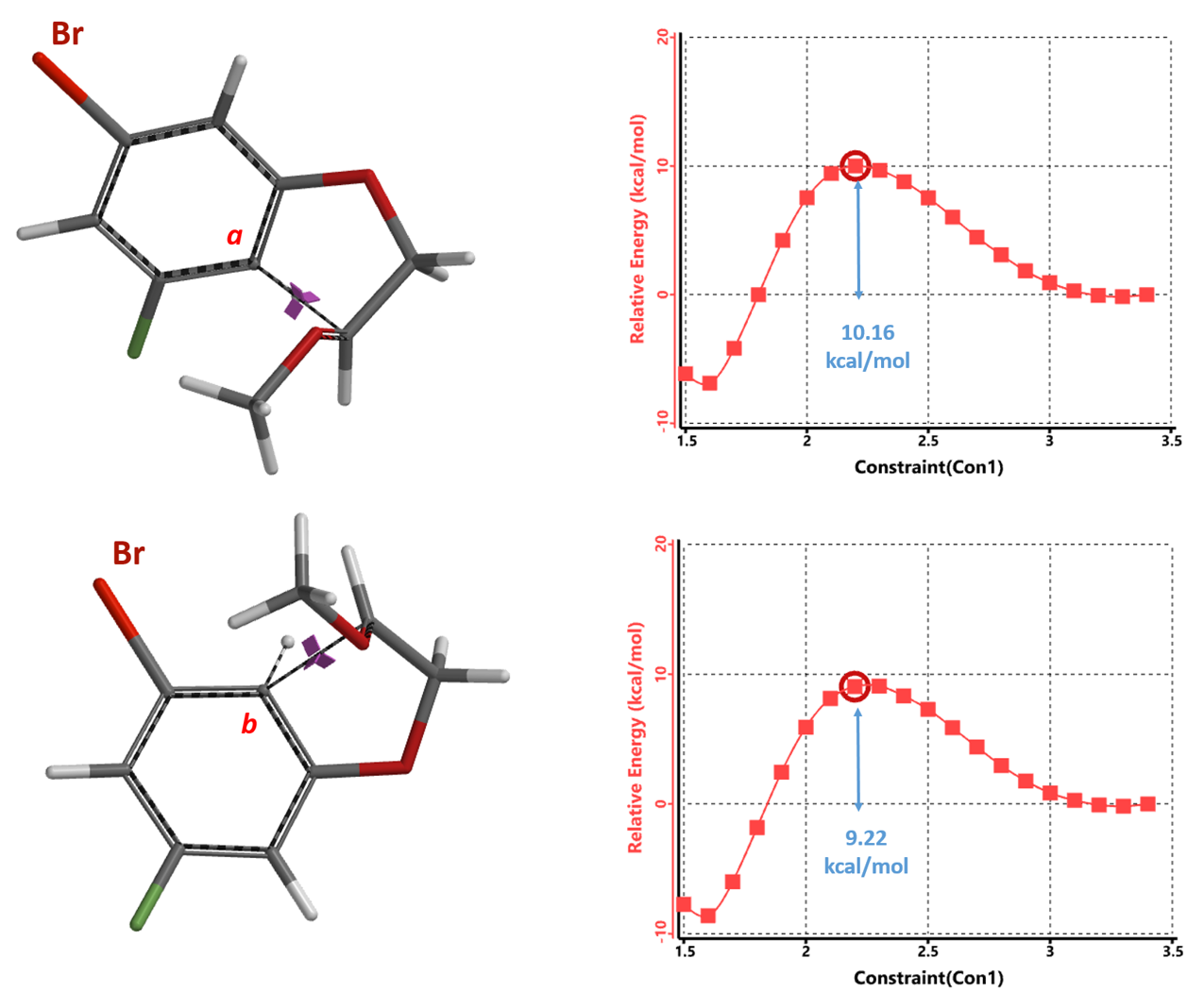
Based on the Arrhenius equation, a 0.94 kcal/mol difference in activation energy at 110 °C translates to a 2a: 2b product ratio of ~1:3.44, consistent with the observed ratio of 1:5.
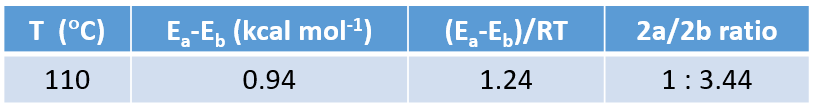
It is worth noting that even though activation energy of the cyclization reaction is ~10 kcal/mol, a reaction temperature of 110 °C is required, presumably for conversion of substrate 1 to oxonium ion A.
In organic chemistry, mechanistic insights focus our QM analyses on crucial structures/factors that determine reactivities observed. What to calculate, How to calculate, How to interpret/question the results. “To ask the right question is already half the solution of a problem.”-Carl Jung
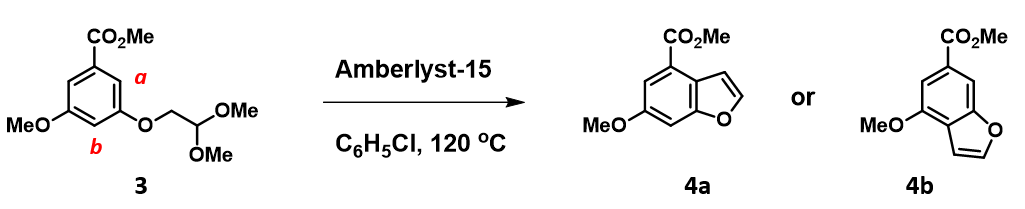
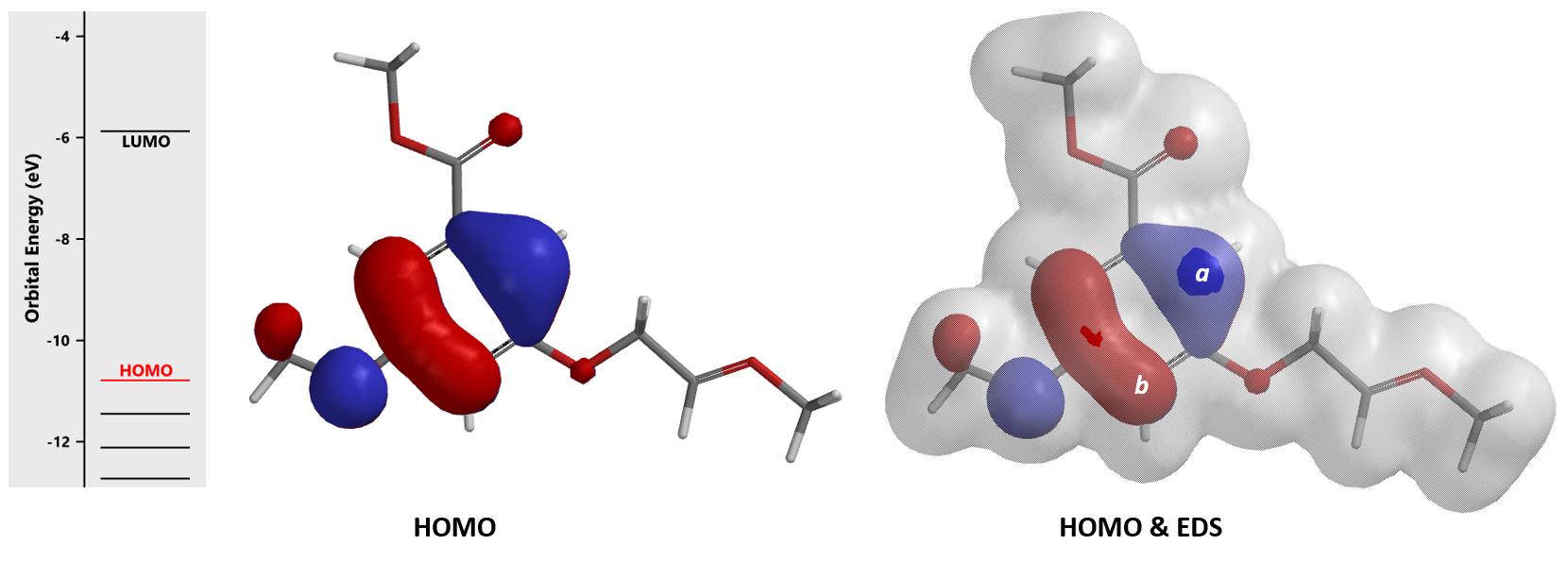
Acid catalyzed cyclization of acetal 3 is reported to be highly regioselective[4]. Since HOMO and HOMO-1 of corresponding oxonium ion have a significant energy gap of 0.66 eV, we could focus QM analysis on the overlay of HOMO and EDS of the oxonium ion (Figure 8). Is 4a or 4b the major product observed?
This article is written and edited by Qiuyue Wang, Zhong Zheng, Shouliang Wang, Yongsheng Chen, John S. Wai
References:
[1] R.J. Nevagia, S.N. Dighe, S.N. Dighe, Eur. J. Med. Chem. 2015, 97, 561.
[2] A. Lavanya, K. Narasimhan, V. Padmini, Mini-Rev. Org. Chem. 2019, 17, 224.
[3] Spartan’20 Tutorial and User’s Guide (2020). Irvine, CA, USA: Wavefunction, Inc. p368.
Electron Density Surface (EDS) that contains >99% of a molecule’s electrons roughly corresponds to a space-filling model, that is, a van der Waals surface. EDS reveals overall molecular size and shape and demarks the steric barrier seen by encroaching molecules, and could be overlaid with molecular orbitals and mapped with electrostatic potential to rationalize physical and chemical properties of molecules.
[4] J-T. Liu, T.J. Do, C.J. Simmons, J.C. Lynch, W. Gu, Z.X. Ma, W. Xu, W.P. Tang, Org. Biomol. Chem. 2016, 14, 8927.