












网站维护
系统内容更新/升级中


Pyrimidines are extensively used in drug discovery due to their unique chemical properties. For medicinal chemists, functionalization of the pyrimidine ring with nucleophilic aromatic substitution reaction (SNAr) of properly substituted 2,4-dichloropyrimidines is a very useful synthetic strategy. We learned that nucleophilic substitution of 2,4-dichloropyrimidines is usually C-4 selective. However, often a time, we also encountered exceptions[1][2][3], that is, C-2 selective displacement or a mixture of products. In this chapter, we continue our QM analyses on why regioselectivity of SNAr reactions observed with 2,4-dichloropyrimidines is so sensitive to other substitutions on the ring.
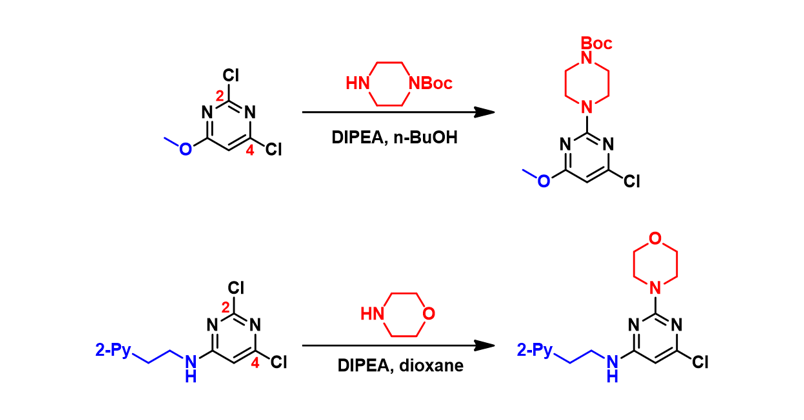
We selected the above two reactions for QM analysis (Fig. 1). For 2,4-dichloropyrimidine with an electron-donating substituent at C-6, its SNAr reactions proceed preferably at C-2, instead of C-4. Authors of the second reaction verified the structure of the product by hydrogenating off the chlorine group. Two distinct doublets were observed in the aromatic region of the 1H NMR spectrum, thus enabling accurate structural assignment of the product[2]. Could we use QM analysis to account for such reversal in selectivity?
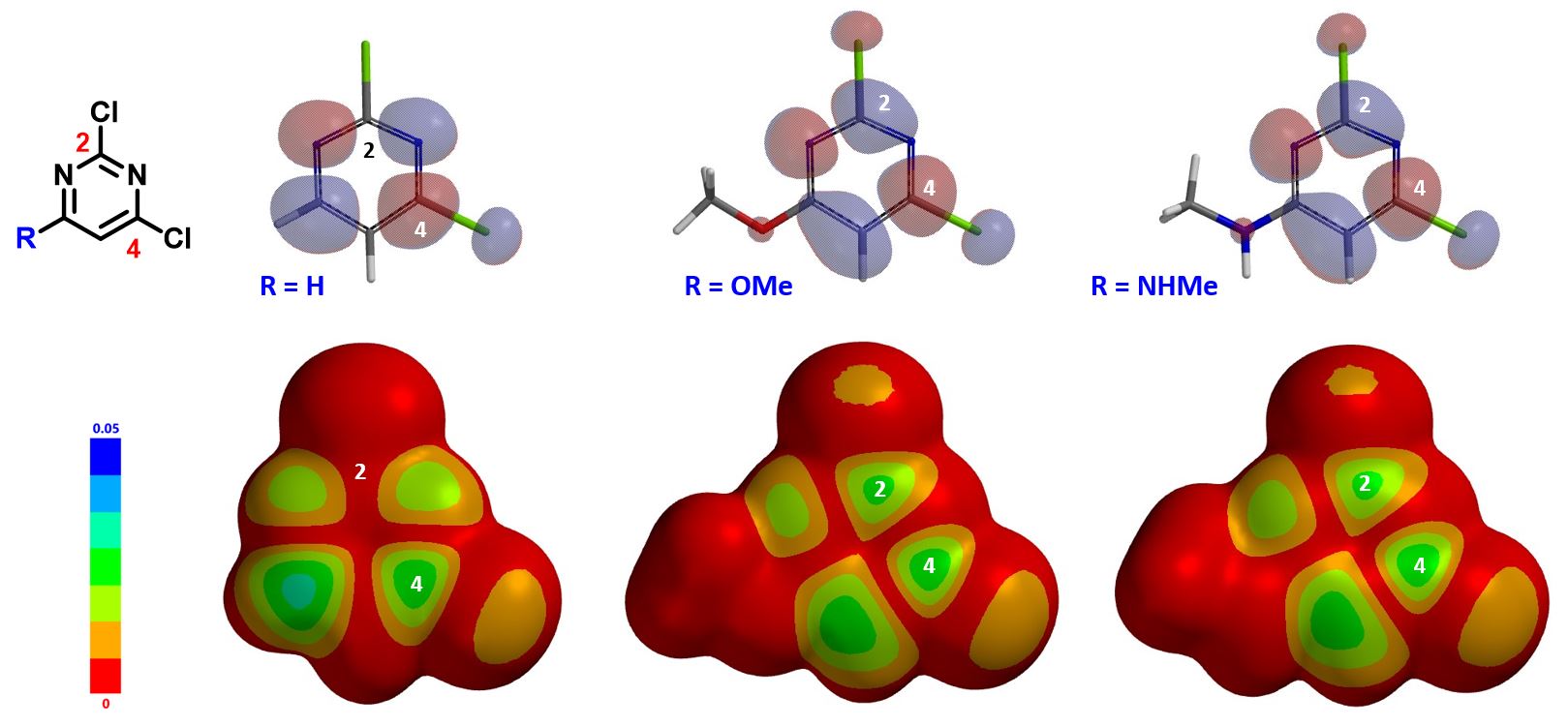
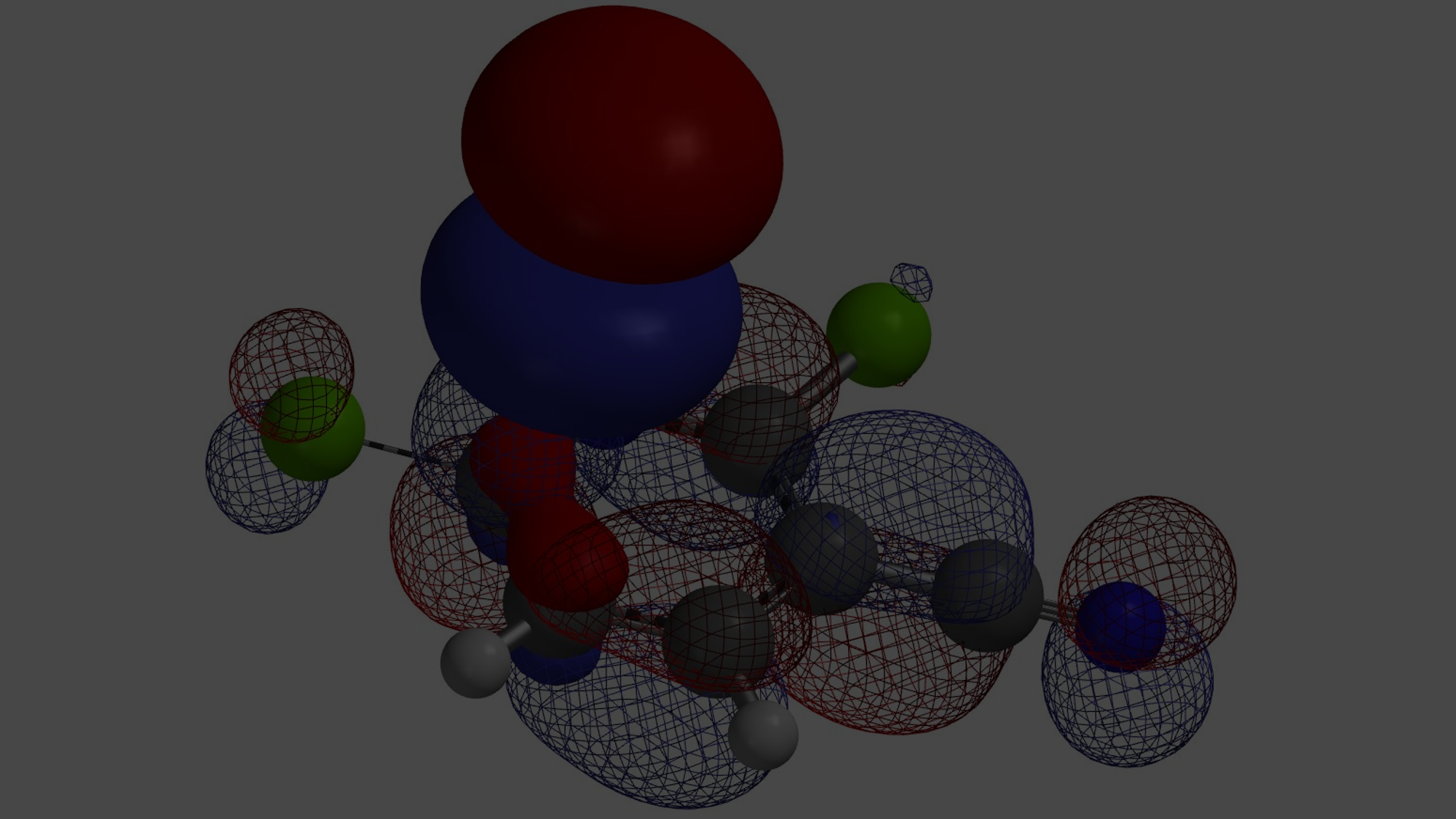
Shown in Figure 2 are LUMOs and LUMO maps of three 2,4-dichloropyrimidines (C-6 R = H, OMe, NHMe) obtained from DFT calculations (Spartan’20, wB97X-D/6-31G*). When R is H, LUMO is mainly distributed at C-4, and almost none at C-2, consistent with what we learned from general heterocyclic chemistry textbooks, accounting for C-4 selectivity observed. On the other hand, when R is OMe or NHMe, LUMOs of the pyrimidines are quite different. LUMO lobes at C-2 and C-4 are similar in size, and on LOMO maps, they have similar concentration, suggesting that we could potentially obtain a mixture of C-2 and C-4 substitution products. However, highly selective C-2 substitution is observed. LUMO and LOMO map analyses could account for increased C-2 reactivity but not the selectivity.
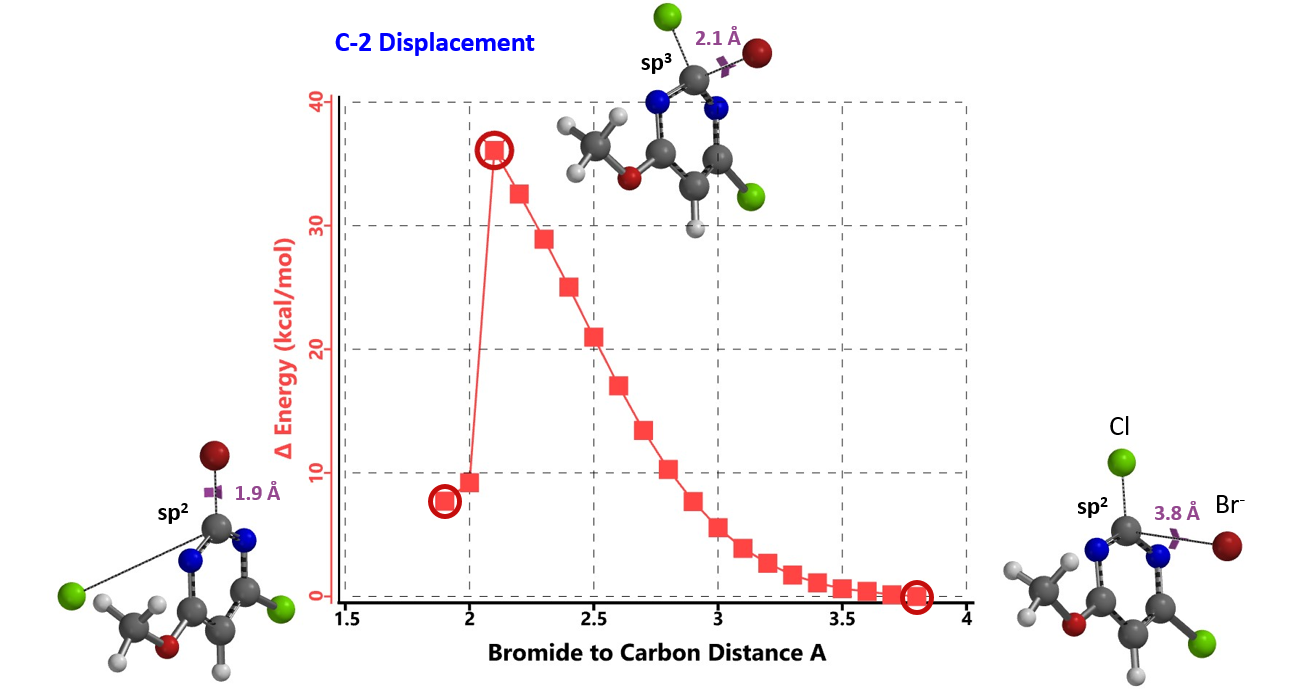
In this case, we calculated for the reaction energy profiles for the C-2 and C-4 substitutions (Figure 3; similar for C-4, not shown), and used bromide as surrogate nucleophile to speed up the calculations[4].
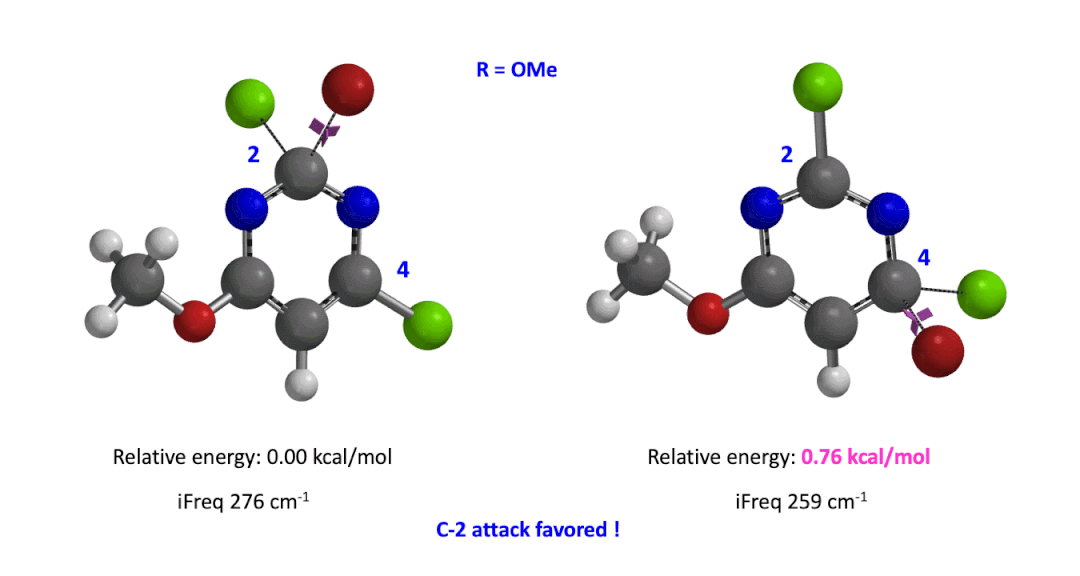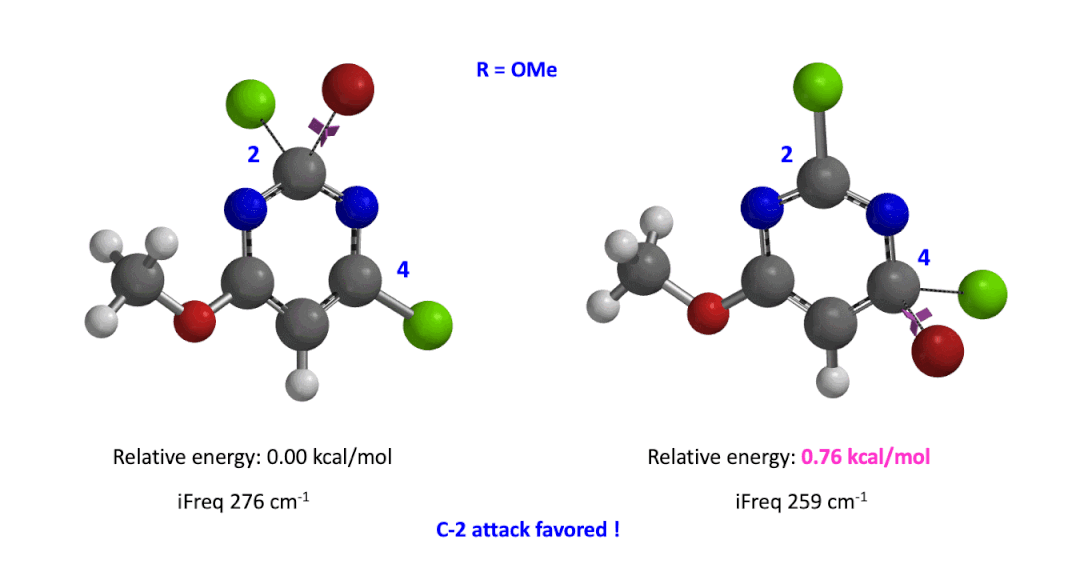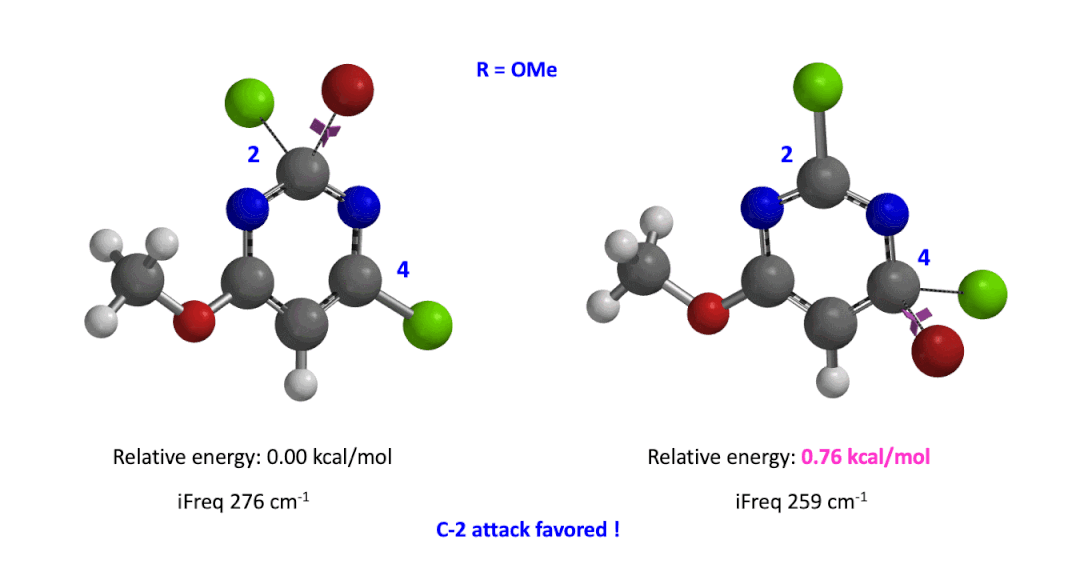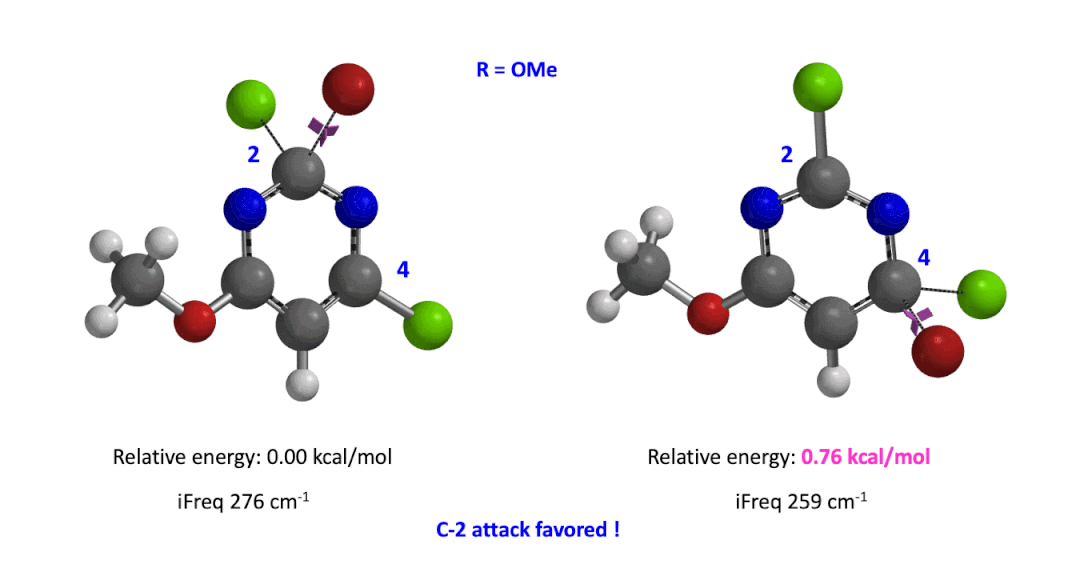
Next we took the structures at the highest points from the two reaction energy profiles to do more accurate “Transition State Geometry” calculations and determine their relative energy difference. Energy of C-4 TS is found to be 0.76 kcal/mol higher than that of C-2 (Figure 4), suggesting that the reaction will be C-2 selective, consistent with experimental observations. Infrared vibration calculation showed one and only one imaginary frequency at 276 and 259 cm-1 for C-2 and C-4 transition states, respectively[5].
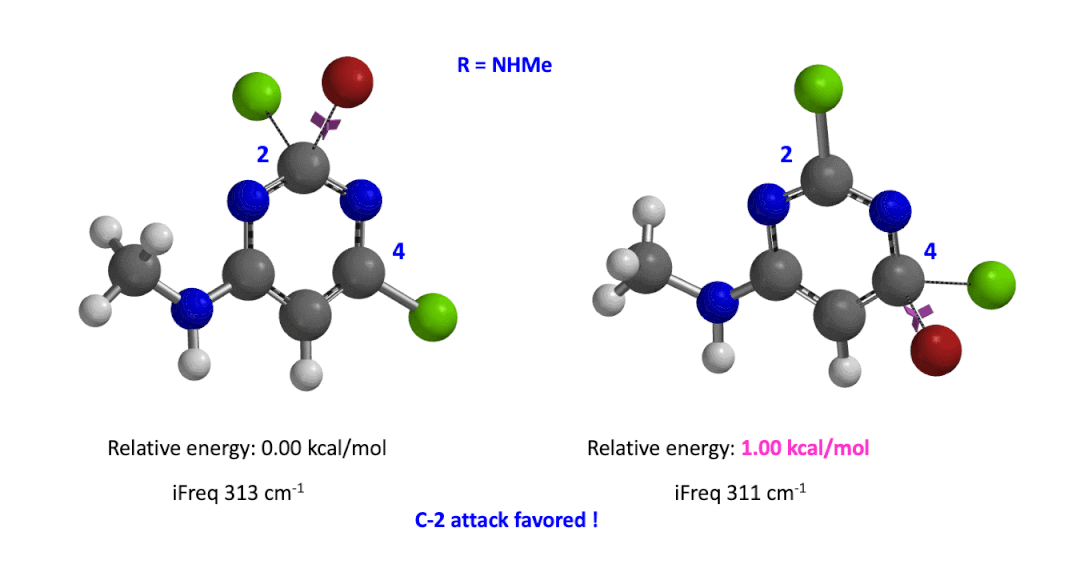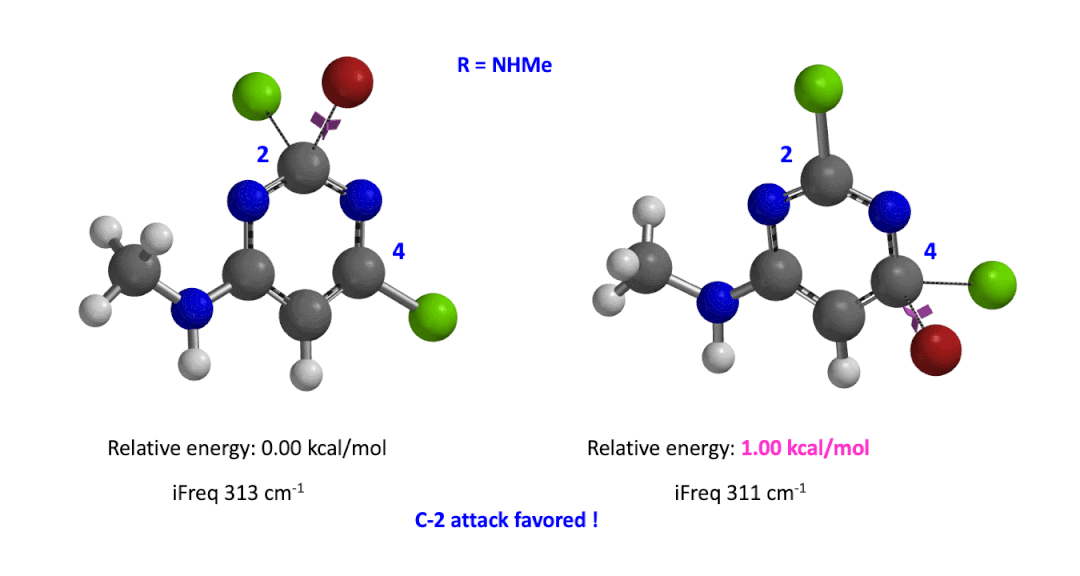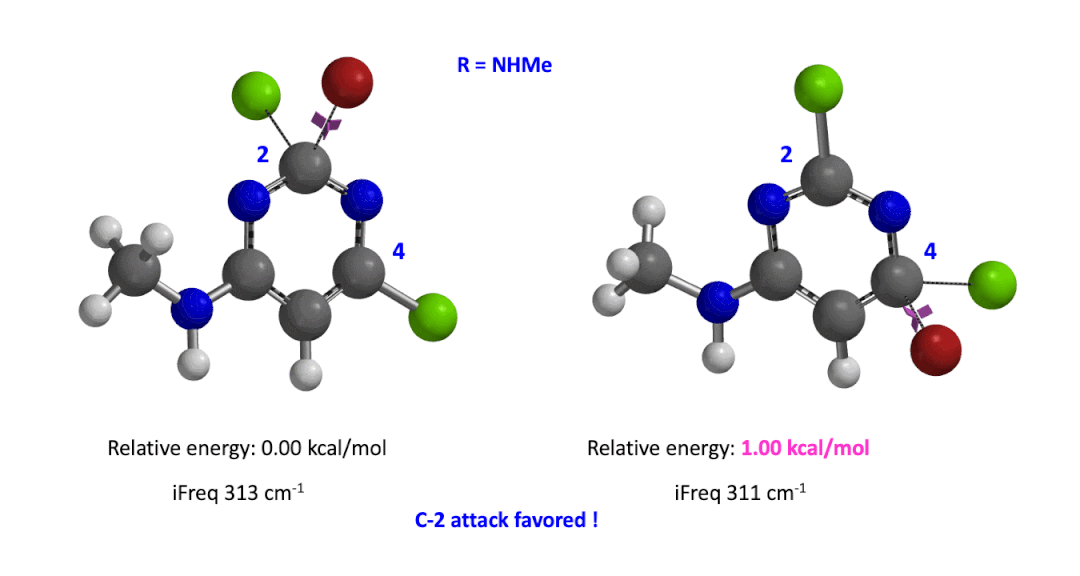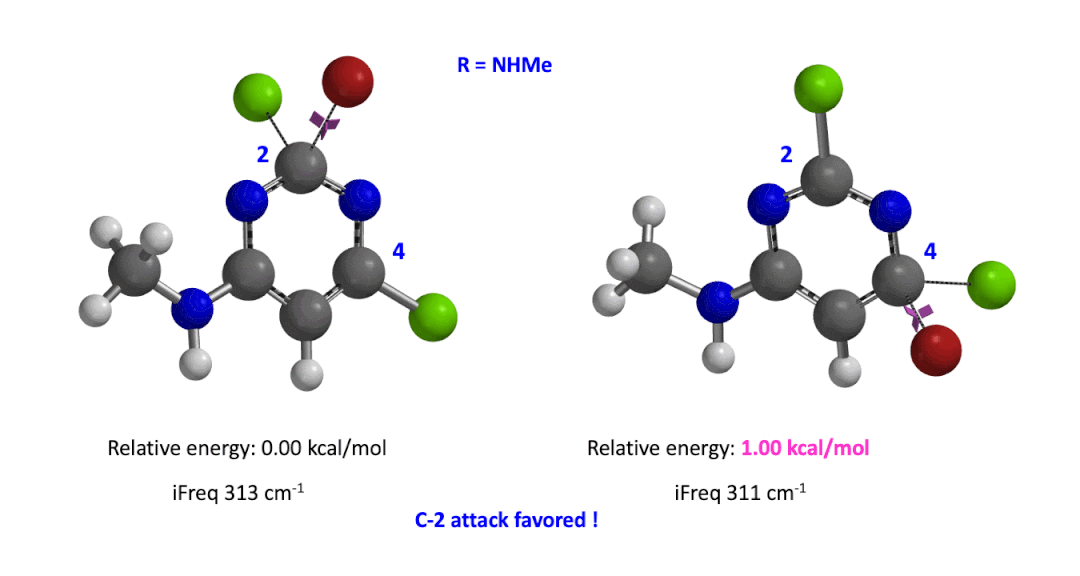
We used the same calculation workflow for R = NHMe to determine their relative energy difference. Energy of C-4 TS is 1.00 kcal/mol higher than that of C-2, suggesting that the reaction at C-2 will also be more favorable (Figure 5).
When we do LUMO analyses for regioselectivity of these competing reactions, we evoked the Hammond hypothesis, that is, in exothermic reaction, the structure of the transition state is similar to the substrates. This has its limitations. When orbital lobes are similar in size, there could be other factors affecting the relative energy of the transition states and altering the product ratio. Under such circumstance, we need to calculate for reaction energy profiles and transition states for proper comparison.
In Chapter 7, LUMO Analysis of Electrophiles (Part II), we mentioned that if the energy difference between LUMO and LUMO+1 is small, we need to consider both orbitals, along with potential steric interaction from neighboring substituents.
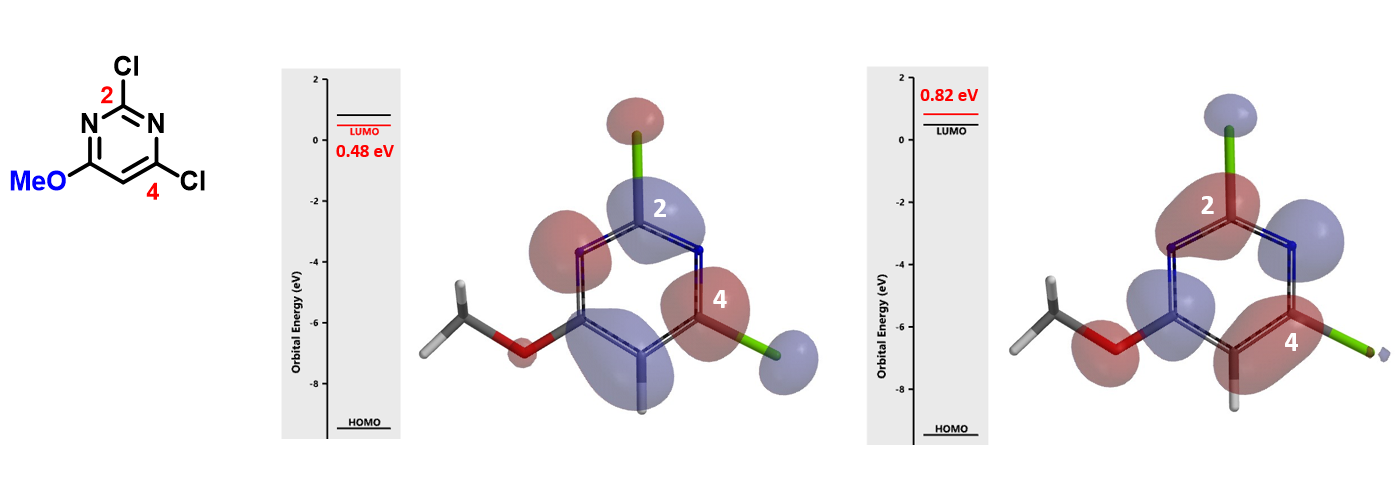
In the case of 2,4-dichloro-6-methoxypyrimidine (Figure 6), the energy difference between LUMO and LUMO+1 is 0.34 eV. With a big enough energy gap, LUMO+1 is not likely to participate in the reaction.
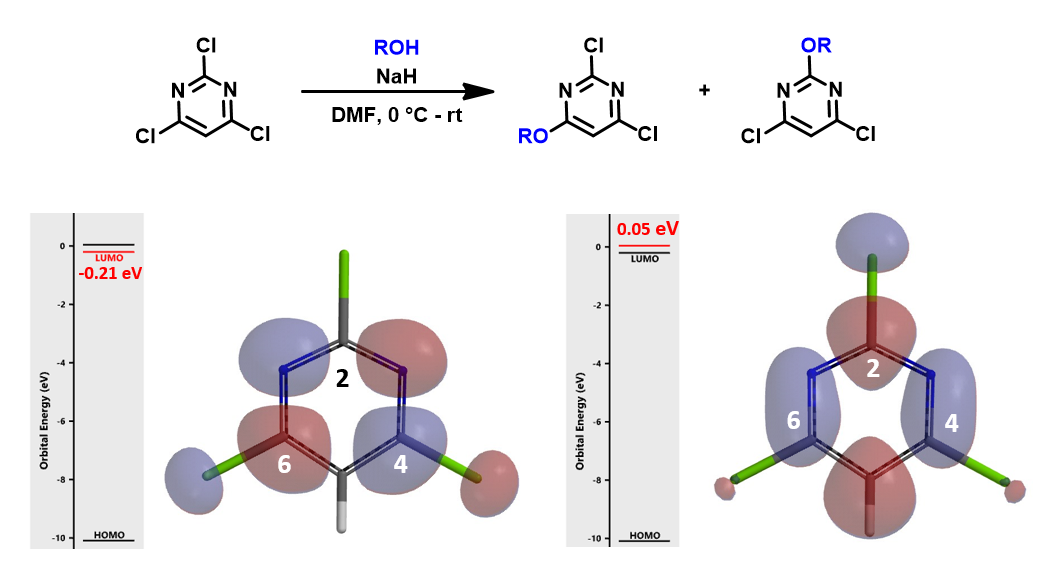
Replacing C-6 methoxy with one more chloro group, we have trichloropyrimidine, which has LUMO lobes on both C-4 and C-6, but no LUMO distribution at C-2 position. Moreover, the energy gap between LUMO and LUMO+1 is 0.25 eV, requiring us to consider LUMO+1 as well. Since there is significant LUMO+1 lobe at C-2, we need to evaluate relative difference in transition states energy. Calculation suggested a mixture of products, consistent with the experimental results.
To summarize what we have learned from chapter 7[3] and this chapter, 1) For SNAr reaction with 2,4-dichloropyrimidine analogs, when there are strong electron donating or electron withdrawing groups at positions C-5 or C-6, the reaction may not always occur selectively at the C-4 position. We could potentially obtain a mixture of C-4 and C-2 substitution products, or even C-2 displacement product only; 2) If energy gap between LUMO and LUMO+1 is ≤ 0.25 eV, it is necessary to consider contribution from LUMO+1; 3) When C-5 position is a sterically bulky substituent, it could affect C4/C2 selectivity[3]; 4) Regioselectivity of 2,4-dichloropyrimidine analogs’ SNAr reaction is very sensitive to electronic and steric effects, yet quite predictable with QM calculation tools.
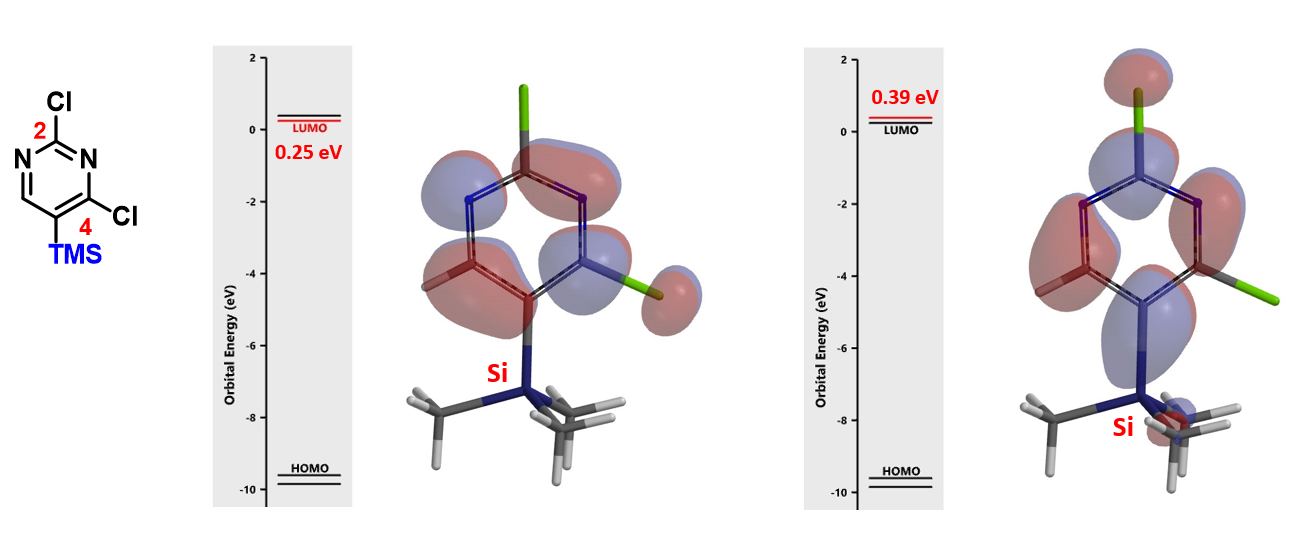
Shown on Figure 8 is 2,4-dichloro-5-trimethylsilylpyrimidines and its LUMO and LUMO+1. energy difference between LUMO and LUMO+1 is 0.14 eV. Only C-2 nucleophilic substitution product could be observed with its SNAr reactions. What could be the reason?
References:
[1] L.D. Arnold, H. Maag, W.W. Turner, Jr. WO 2016/168619 A1.
[2] J.M. Large, J.E. Torr, F.I. Raynaud, P.A. Clarke, A. Hayes, F. Stefano, F. Urban, S.J. Shuttleworth, N. Saghir, P. Sheldrake, P. Workman, E. McDonald, Bioorg. Med. Chem. 2011, 19, 836 – 851.
[3] See Chapter 7, LUMO Analysis of Electrophiles (Part II).
[4] See Chapter 10, SNAr Reaction of Polyhalogenated Heterocycles.
[5] a) Spartan'20 Tutorial and User's Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp. 158, 442, 459 & 536. b) See Chapters 22 and 24 for transition state and imaginary frequency calculations.
This article is written and edited by Renwei Zhang, Liting Dong, Tommy Lai, Yongsheng Chen, John S. Wai