












网站维护
系统内容更新/升级中


.png)
In Chapter 9, we discussed the application of QM to calculate for the difference in activation energy of competing alkylation paths of pyrazole, estimate product ratio, and use the result to guide our synthetic planning and execution.
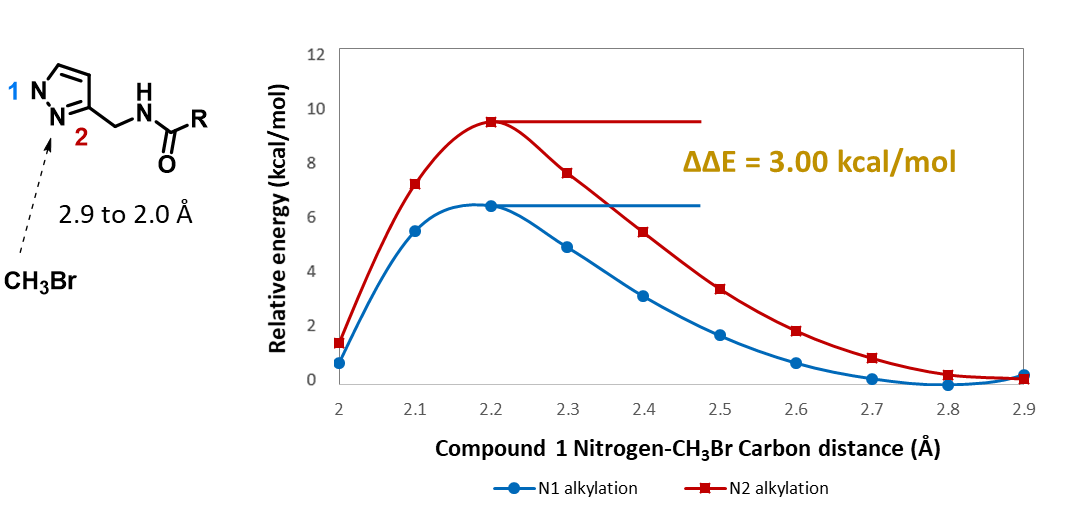
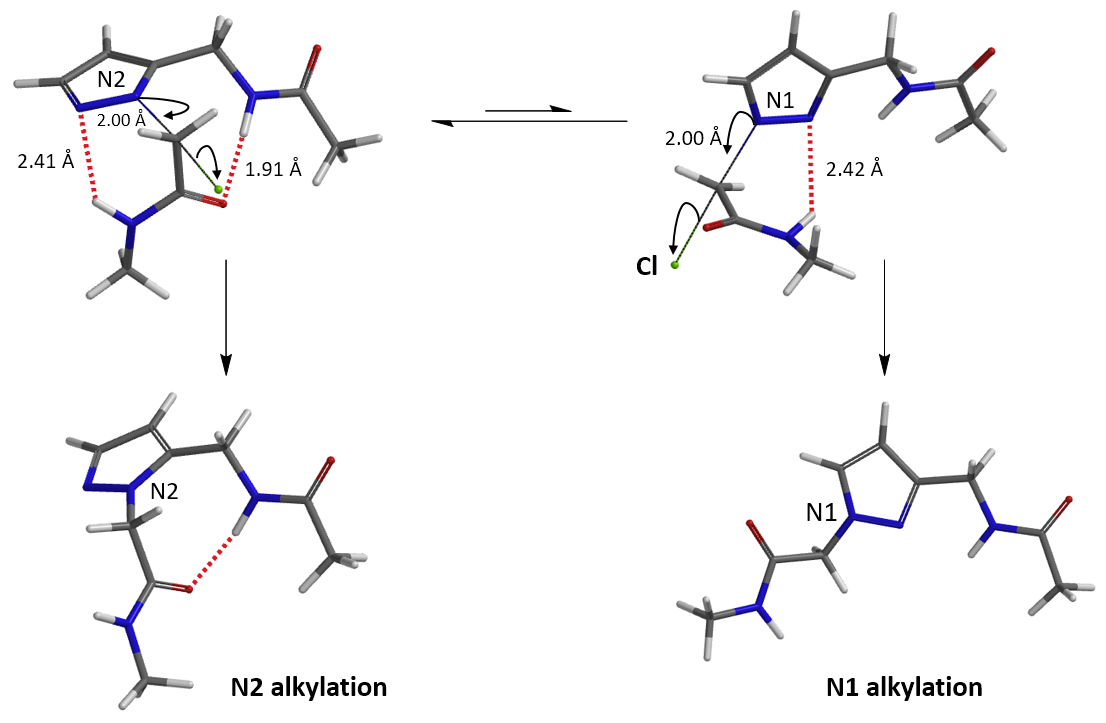
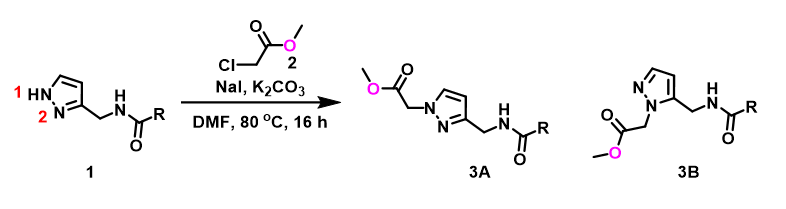
For assessment of competing alkylation of pyrazole 1 (Figure 1), we usually set up two calculations in parallel using CH3Br as generic alkylating reagent to speed up the process: one approaches N1, and the other interacts with N2, to estimate their activation energies for comparison. In this case, the calculated activation energies are 6.4 kcal/mol for N1 and 9.4 kcal/mol for N2 methylation, in favor of N1 alkylation (Figure 2).
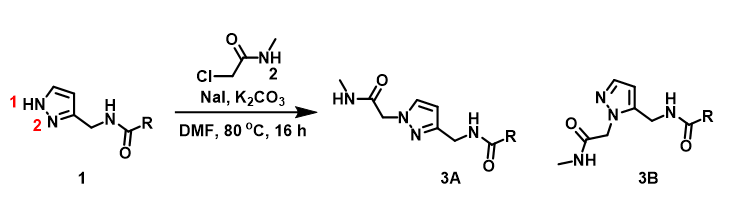
With N-methyl chloroacetamide used as alkylating reagent in our synthesis, only N2 alkylation product 3B was observed, different from what the calculations with methyl bromide suggested.
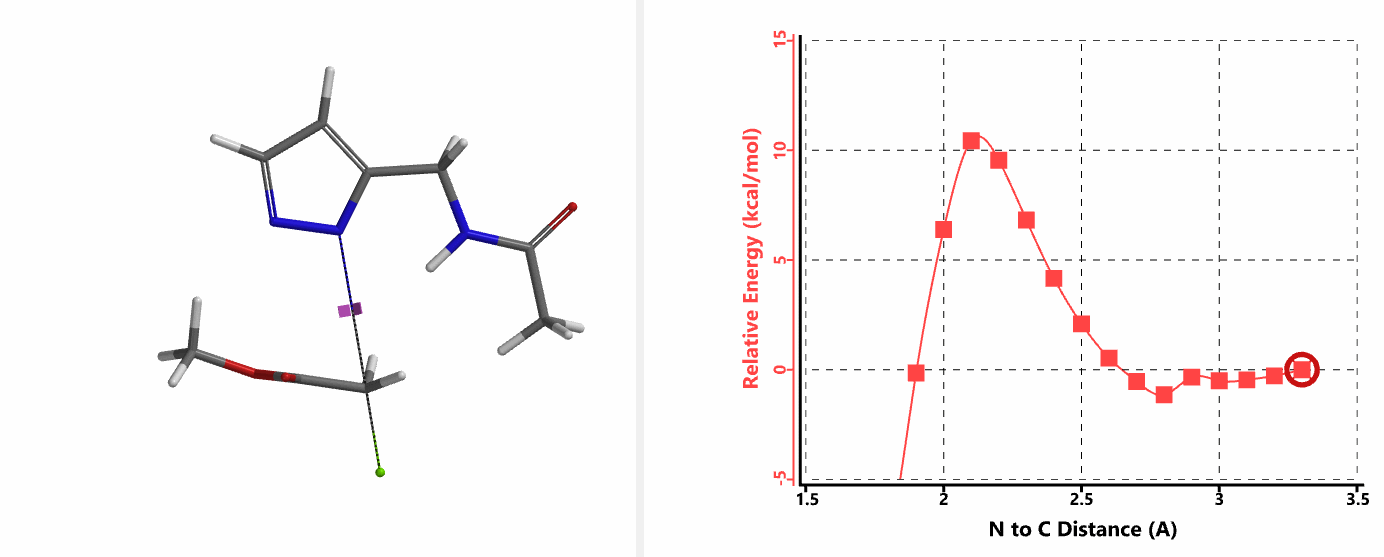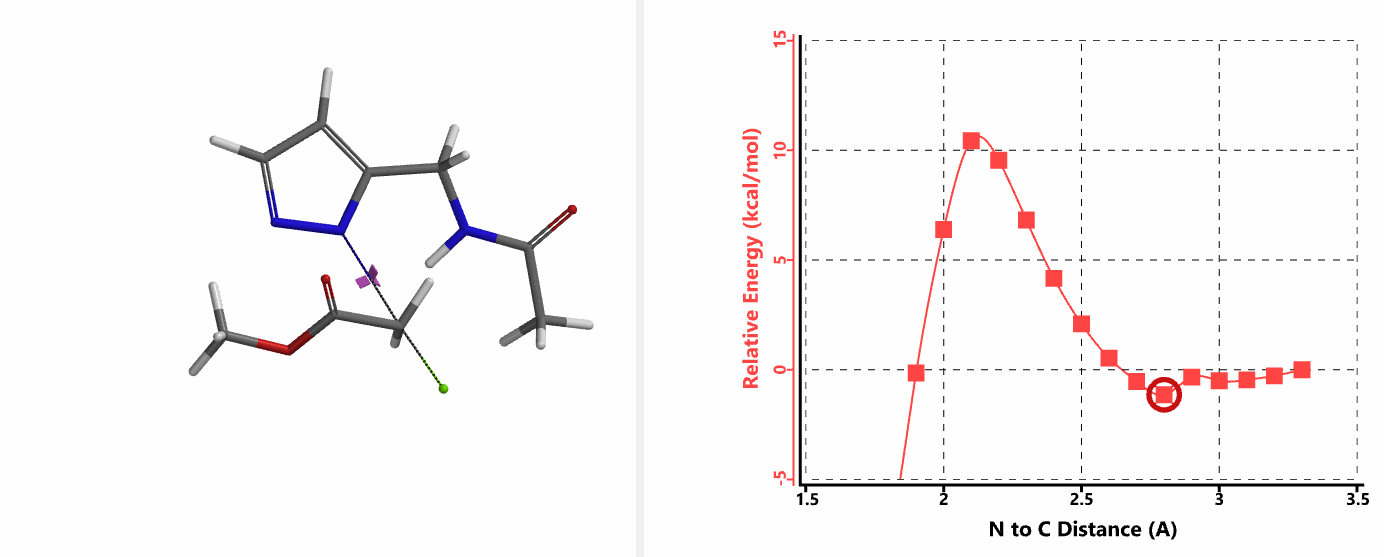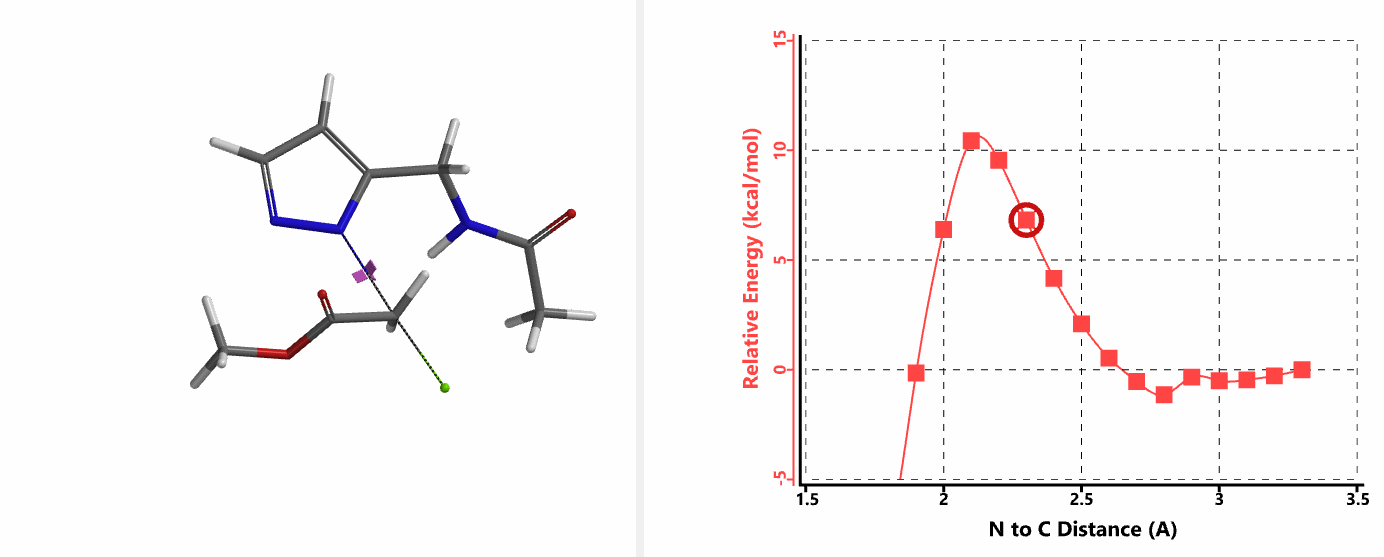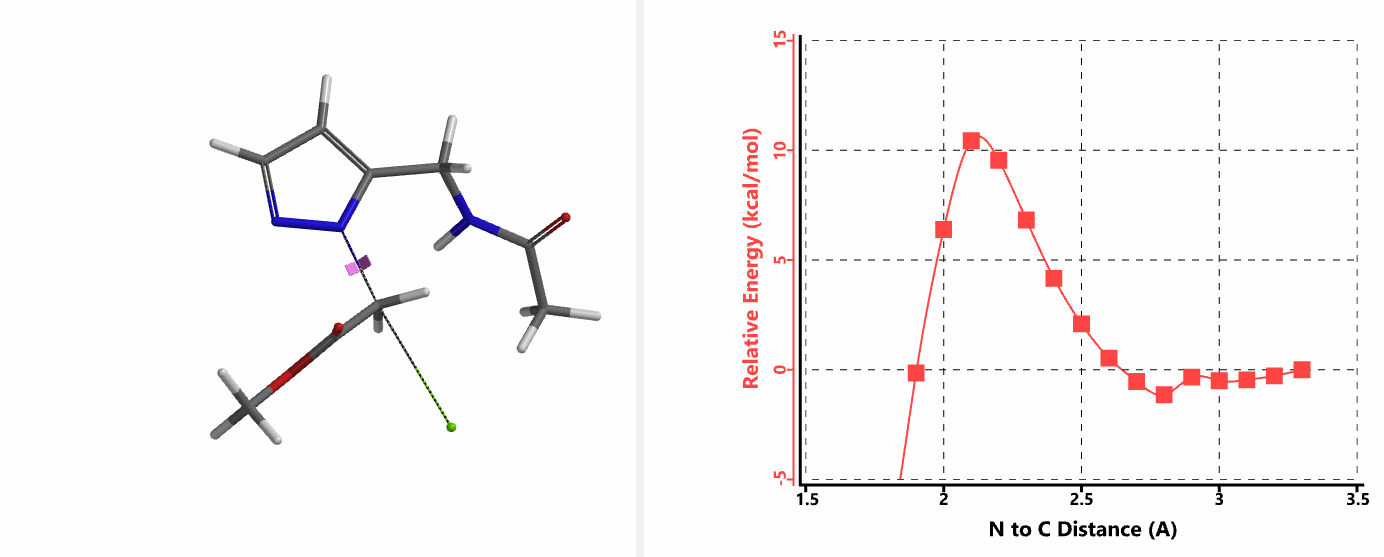
When we repeated the calculations with the chloroacetamide, the activation energy estimated is 18.0 kcal/mol for N1 methylation and 15.0 kcal/mol for N2 alkylation, suggesting a reversal in N1 vs N2 selectivity versus the one modeled with methyl bromide. The 3 kcal/mol energy difference translates to an anticipated 3A:3B product ratio of ~1:70 at the reaction temperature of 80 ºC, consistent with experimental observations (Figure 3). What could have caused a change in N1 vs N2 selectivity?
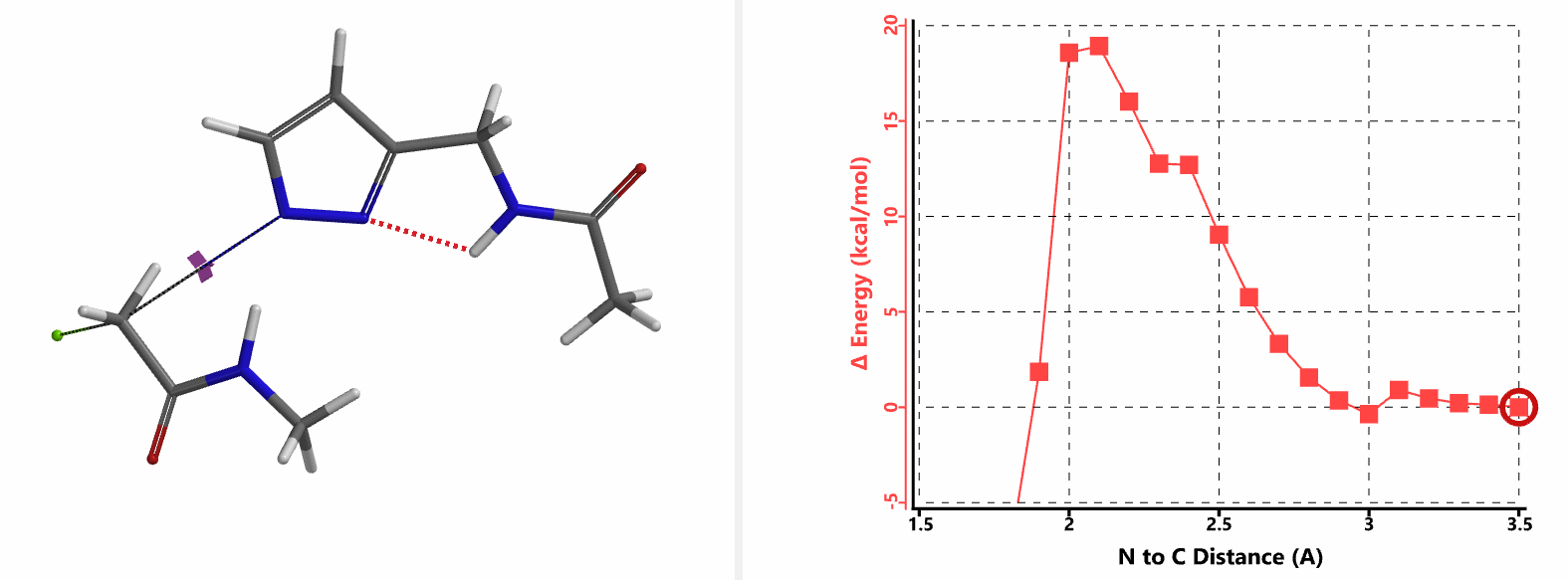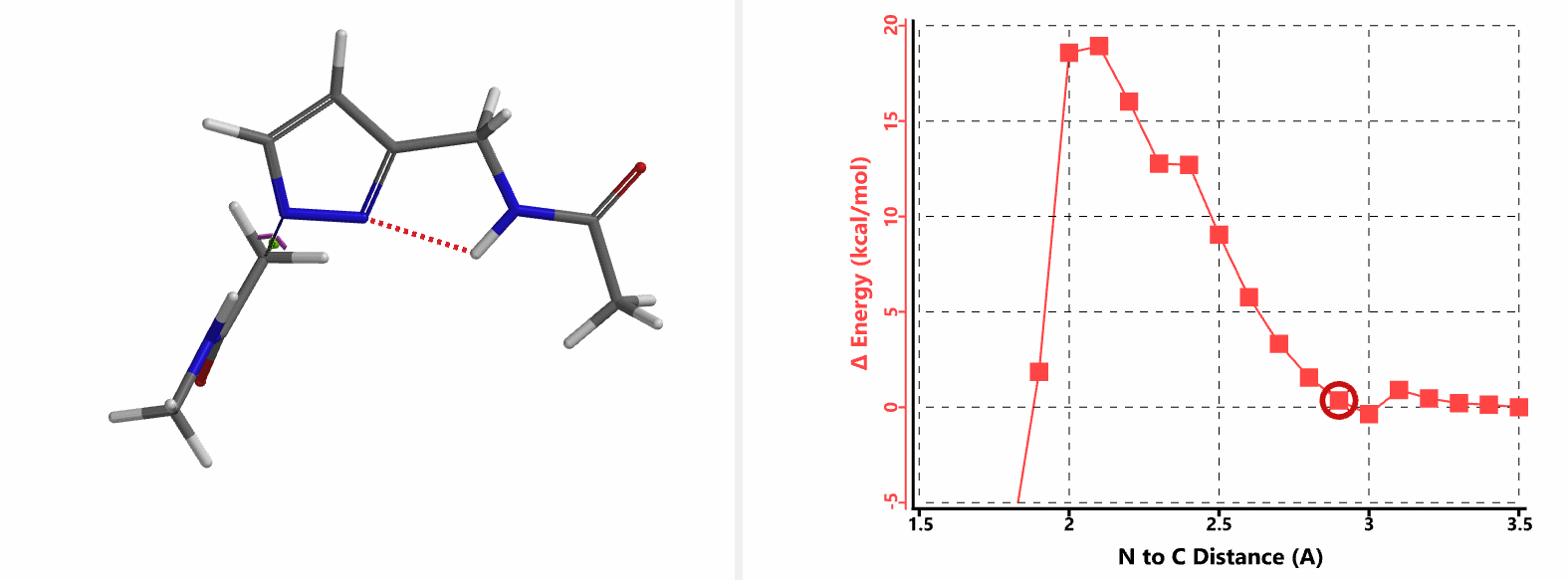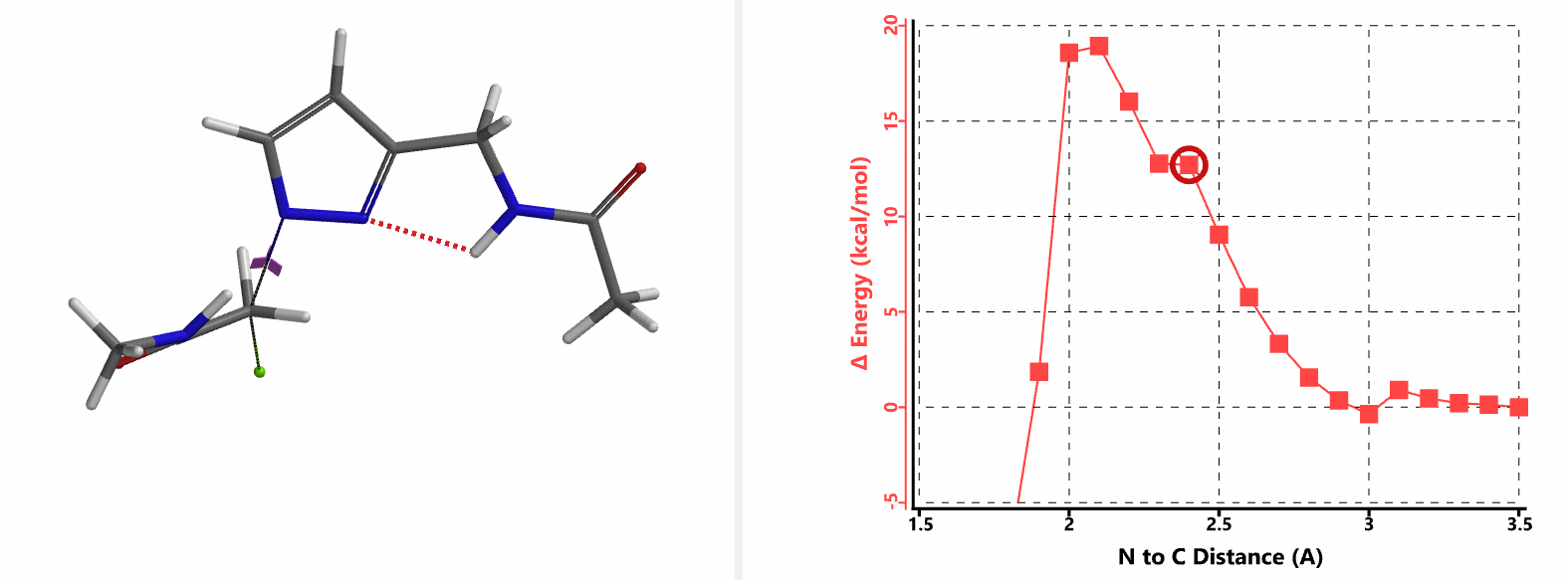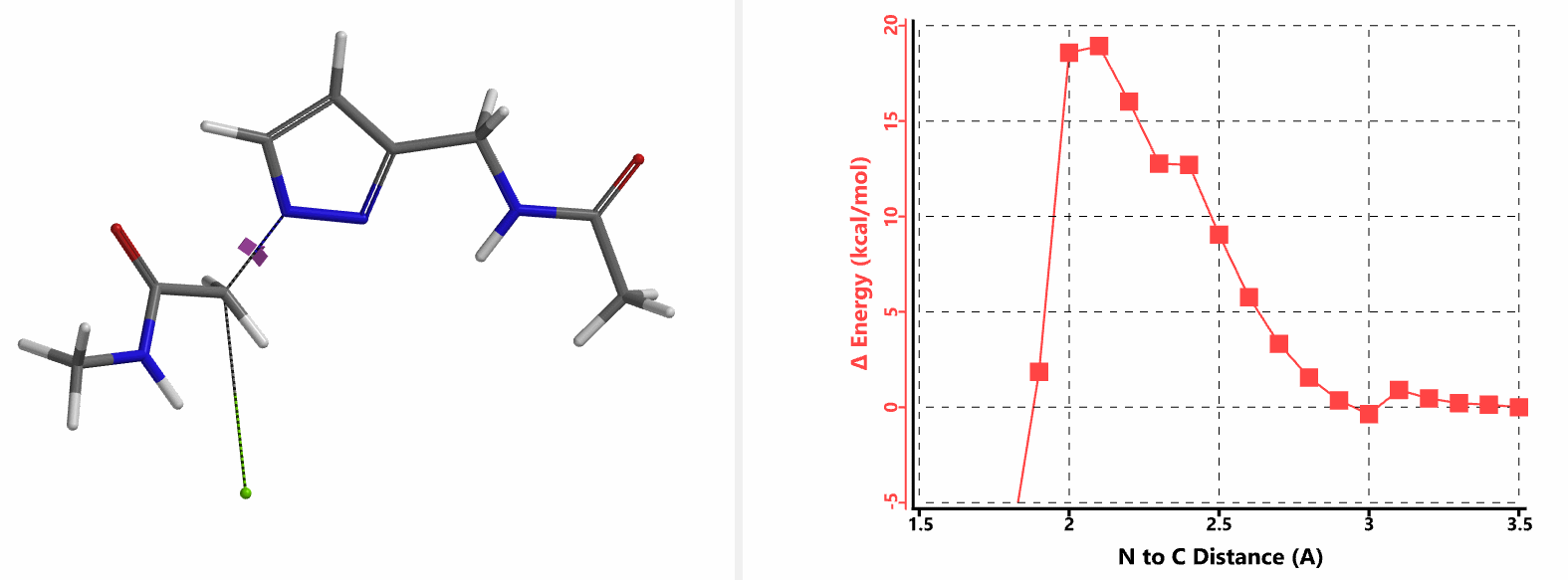
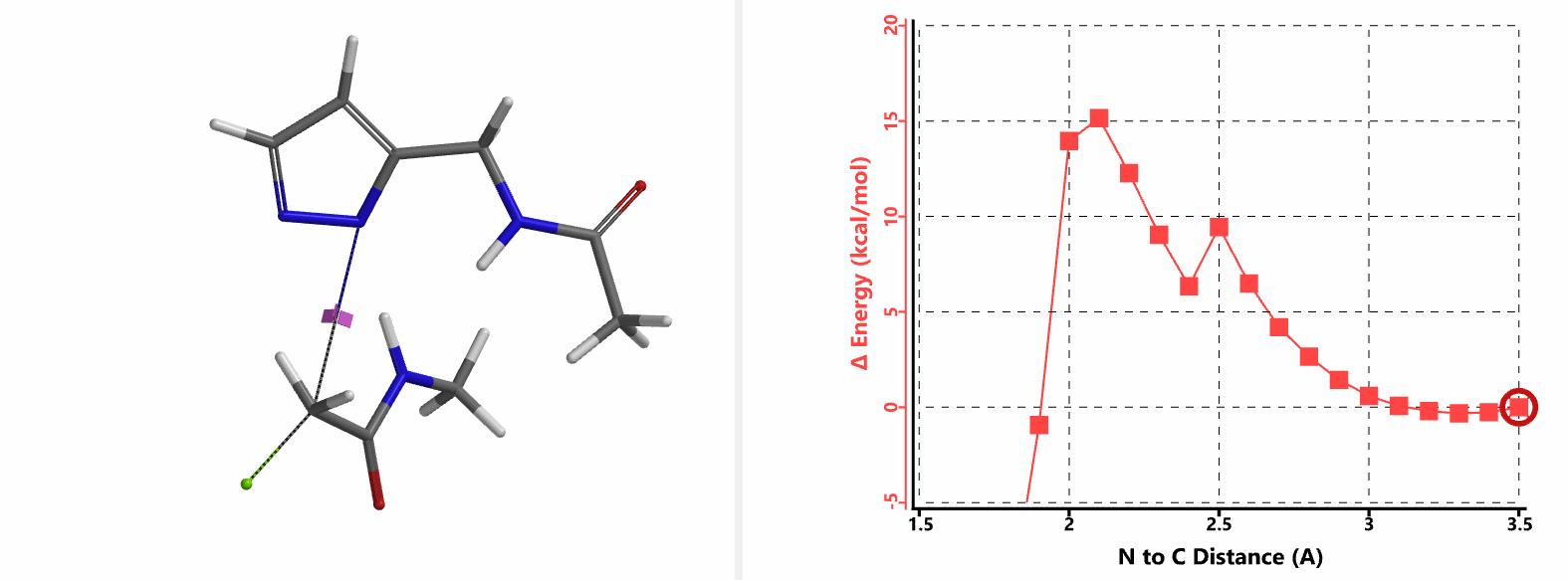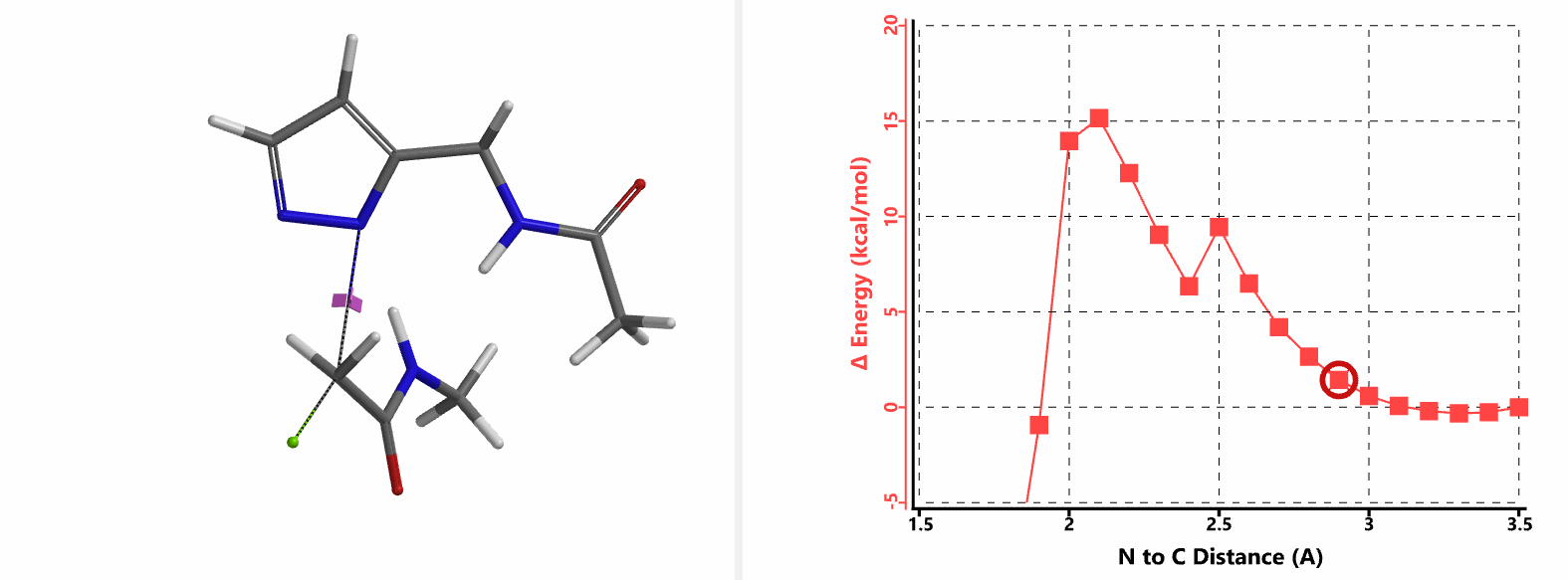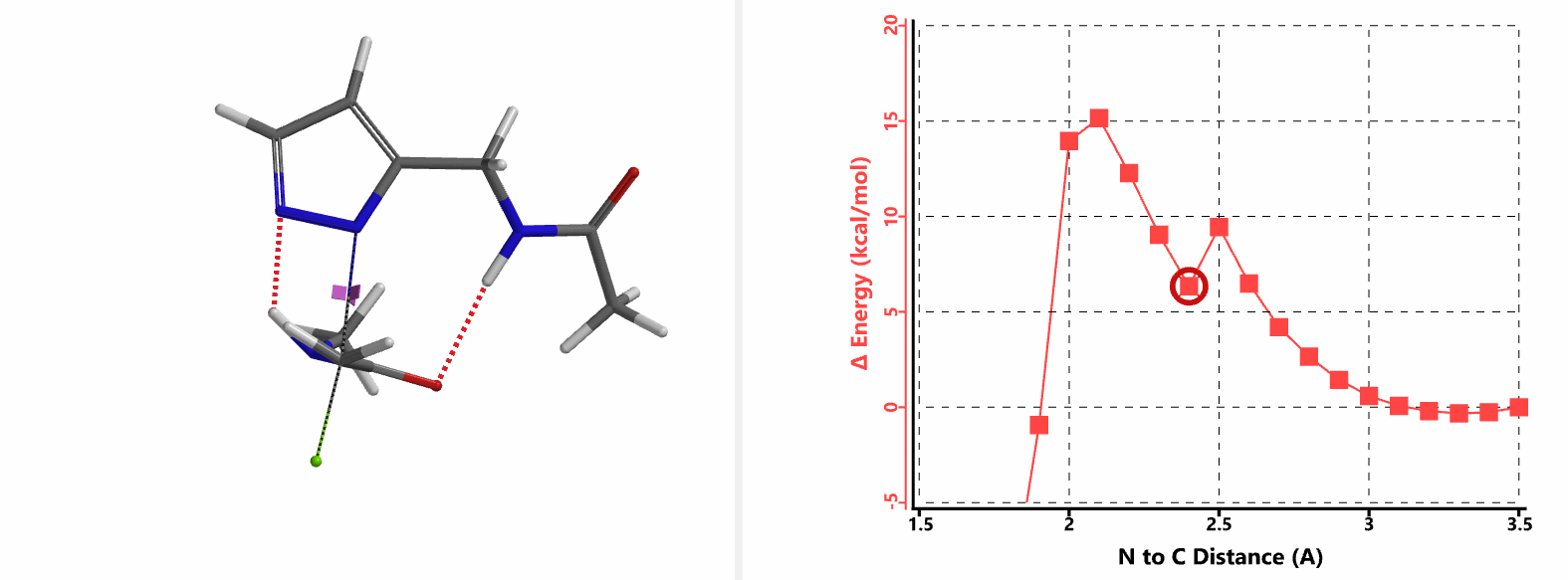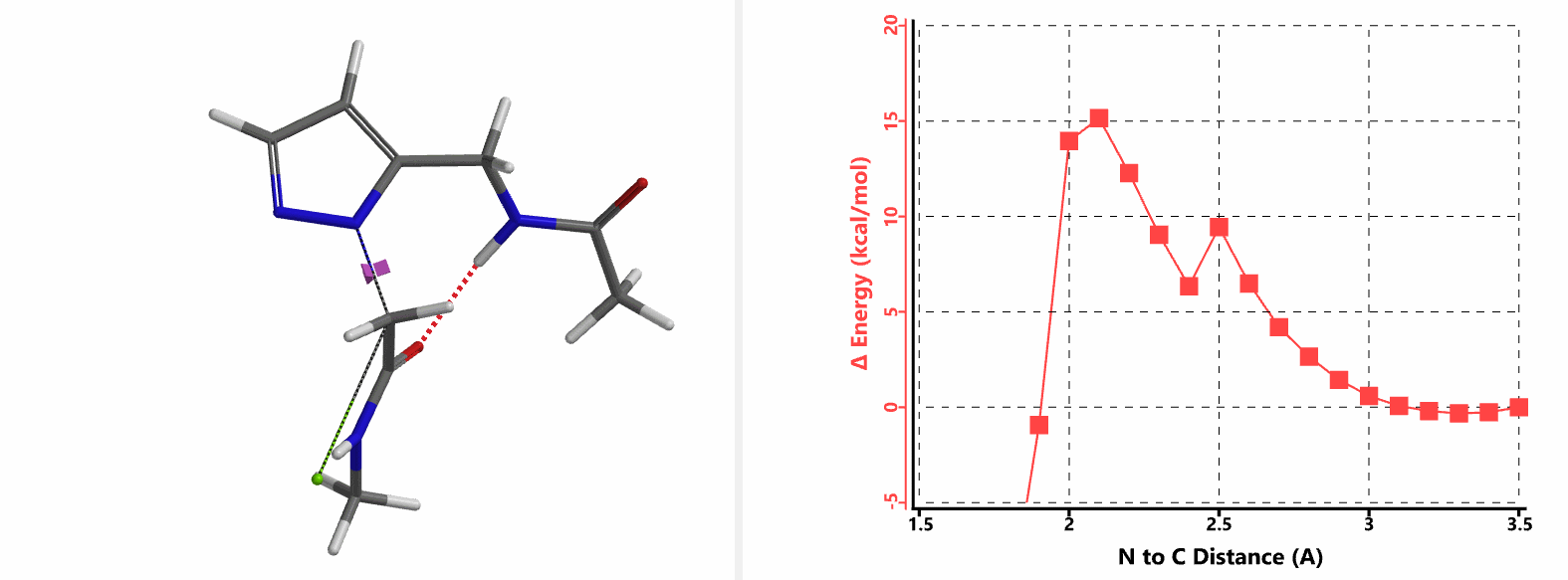
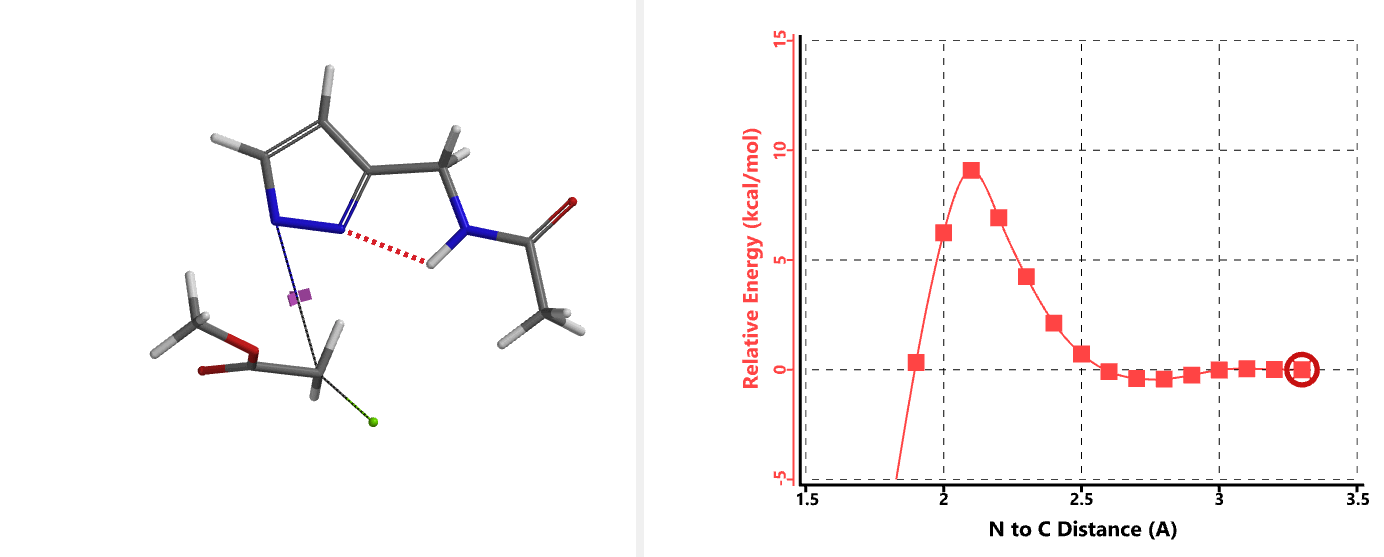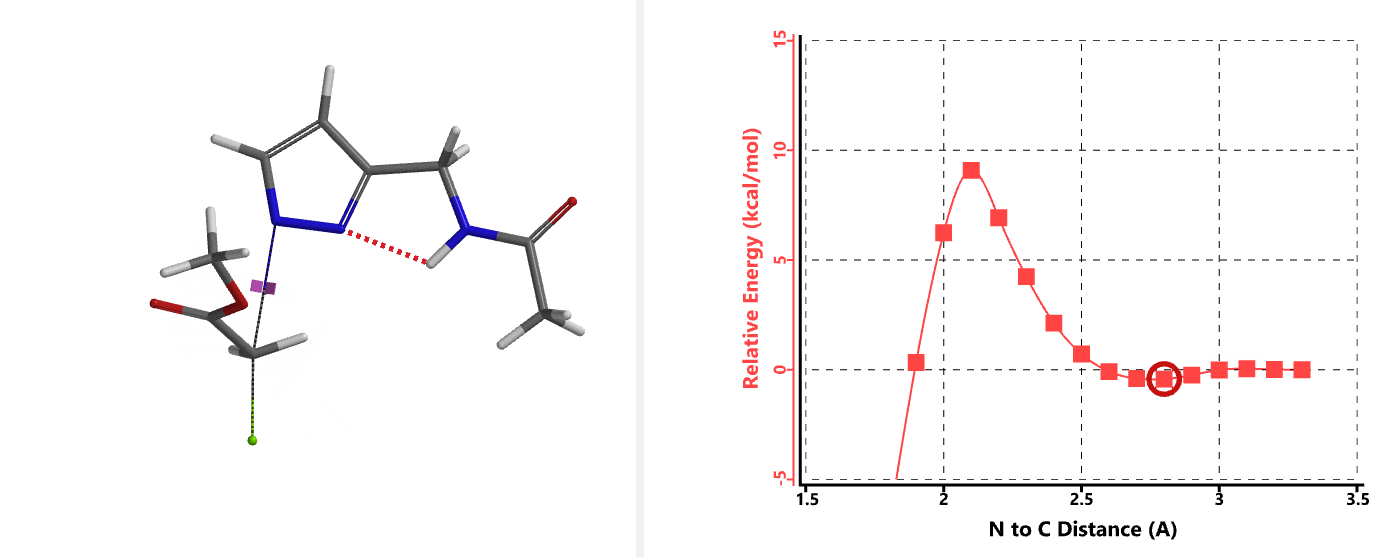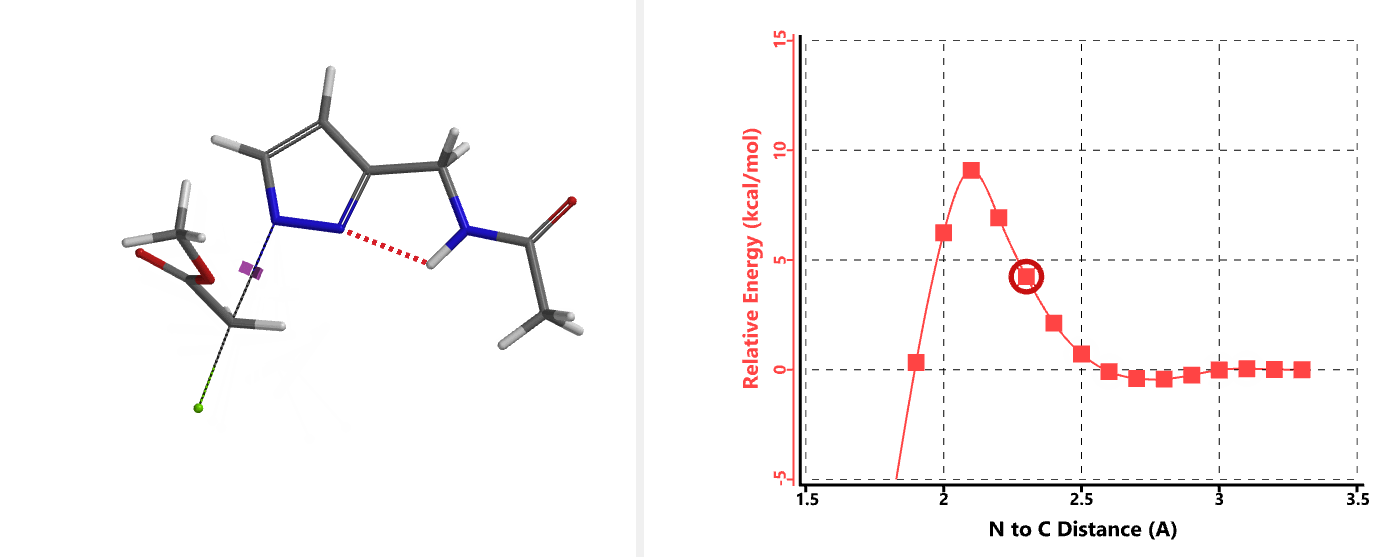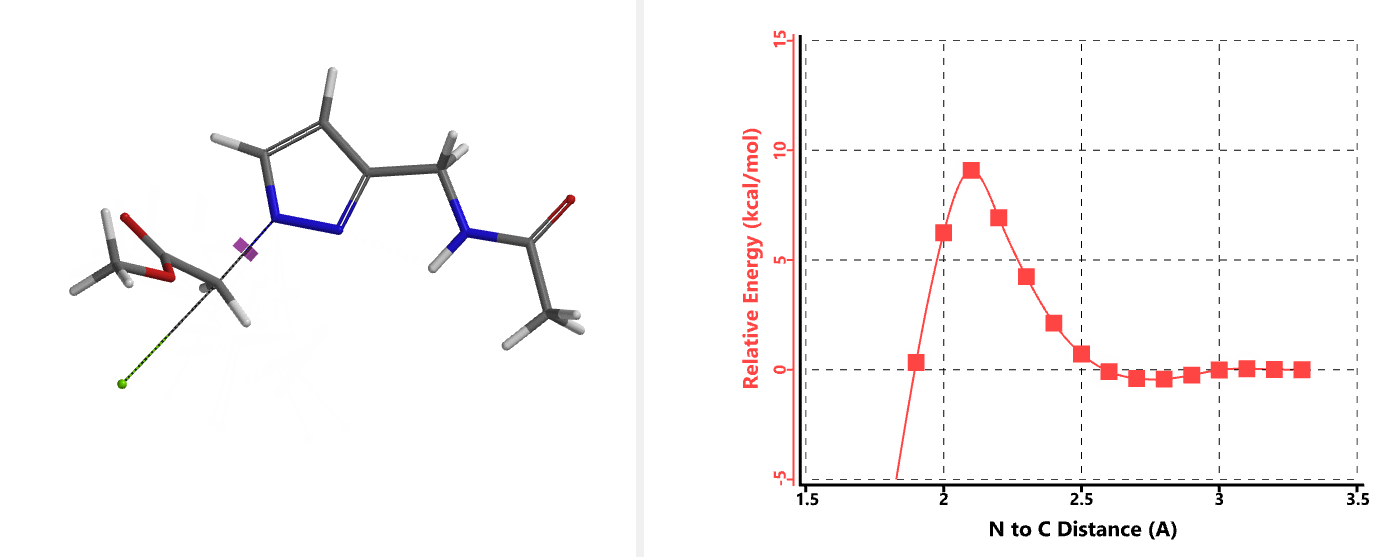
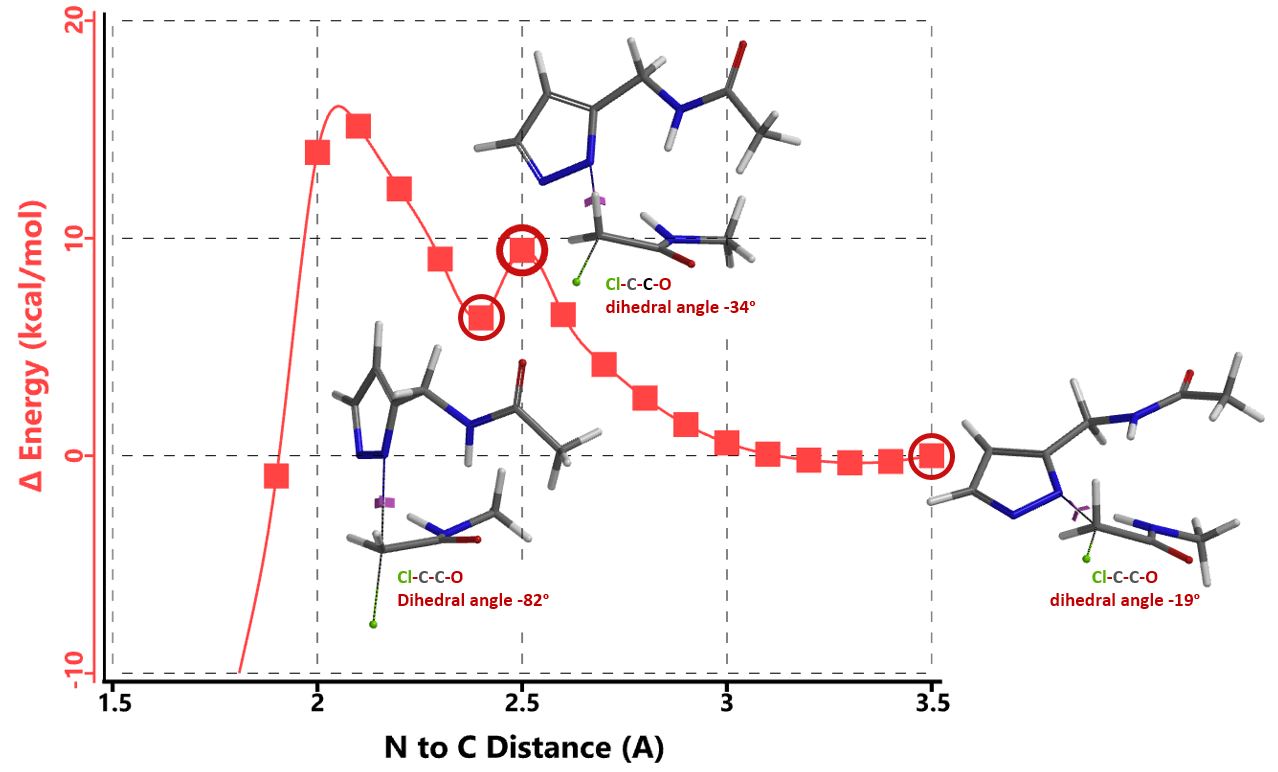
In the calculated transition state structure of N1 alkylation, a hydrogen bond (2.42 Å) between chloroacetamide NH and the pyrazole N2[2] is observed. While for the transition state structure of N2 alkylation, in addition to the comparable hydrogen bond (2.41 Å) between chloroacetamide NH and the pyrazole N1, a second, stronger, hydrogen bond (1.91 Å) is observed between the side chain amide NH and the carbonyl oxygen on the alkylating reagent (Figures 4 and 5). The latter hydrogen bond offers further stabilization to the transition state for N2 relative to N1 alkylation, accounting for the N2 selectivity.[3] It is worth noting that both reaction energy profiles show characteristic bumps at N-C distance ~2.5 to 2.3 Å, due to change in Cl-C-C-O dihedral angle of the incoming chloroacetamide, i.e. the chloro group changing from being close to coplanar relative to the amide to close to perpendicular in transition state.[4]
Transition state (Figure 5) calculations showed one and only one imaginary frequency which corresponds to the bonds being made and broken, supporting the use of these structures to account for the selectivity observed.
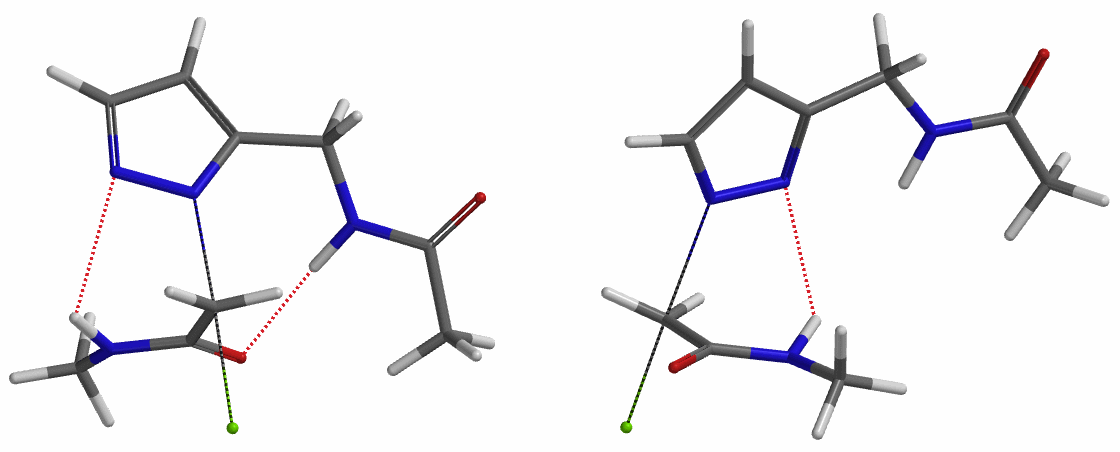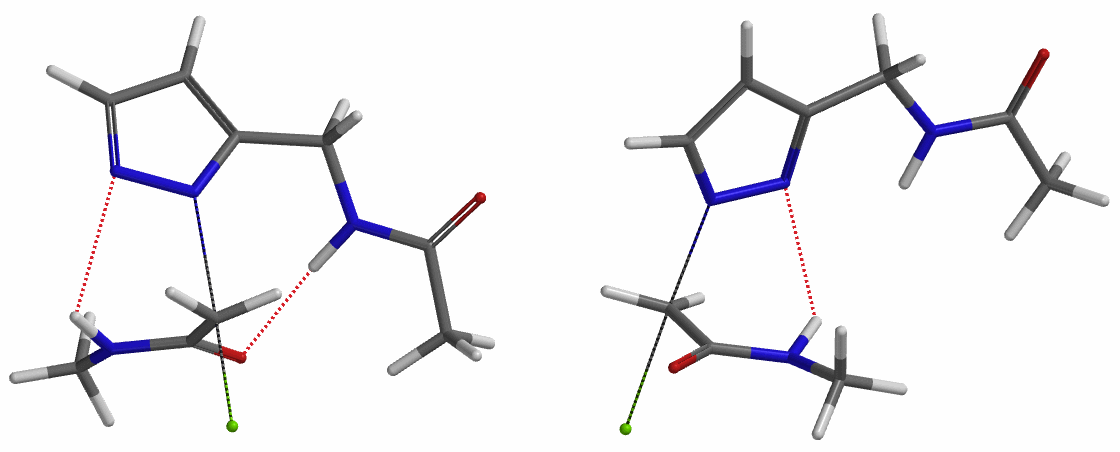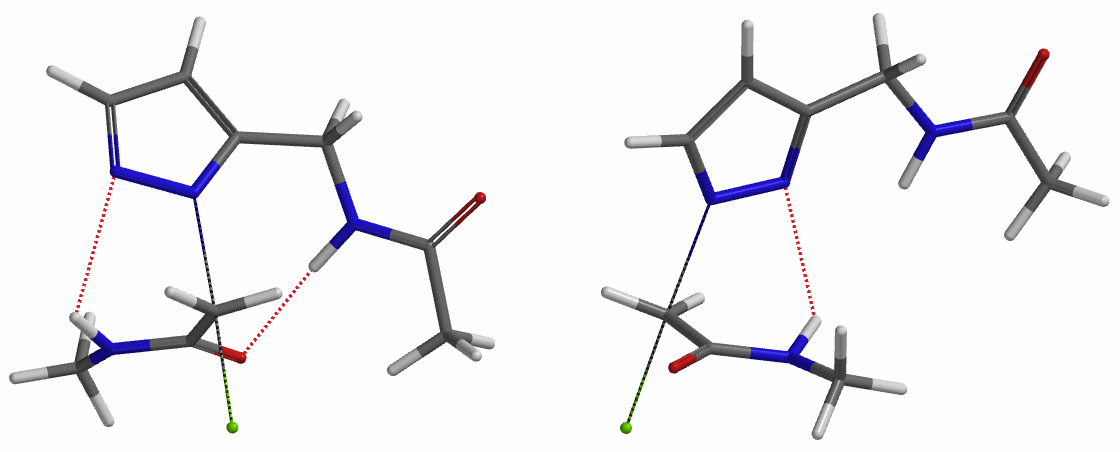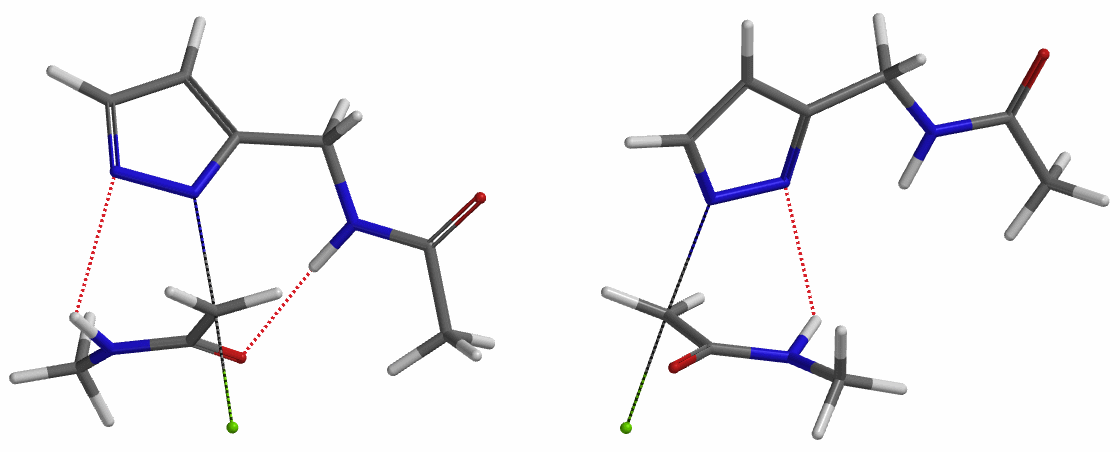
To further evaluate the hypothesis that these hydrogen bonds are indeed key to the N2 selectivity observed with N-methyl chloroacetamide, we replaced it with methyl chloroacetate for calculations. Reaction energy profiles showed little hydrogen bond interaction in the transition states, with estimated activation energy of 9.5 kcal/mol for N1 and 11.6 kcal/mol for N2 alkylation, contrasting to the selectivity observed with chloroacetamide, substantiating our explanation.
The application of relevant computational models for prediction of experimental outcome requires careful consideration of many factors: are surrogate structures representative of the reactions, will there be any intramolecular or intermolecular interactions that are crucial in determining the outcome, do we need to include explicit solvent molecules in the calculations (Chapter 15: Alcohol speeds up Boc protection; Chapter 24: QM study of the reaction mechanism of diazotransfer reaction), etc. Chemical insights do not automatically present themselves as the results of a calculation, but require additional human effort. It is very important that our retrospective QM calculations could account for the experimental results, before they could be considered for prospective analyses.
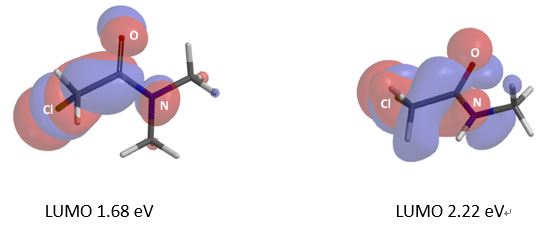
We are interested in understanding the differences in reactivity between thiol and N,N-dimethyl chloroacetamide versus N-methyl chloroacetamide. LUMO and LUMO maps of two amides are shown below in Figure 8. Which one will you expect to react faster with thiol? Hydrogen bond interaction could play a role too? Will it speed up or slow down the reaction?
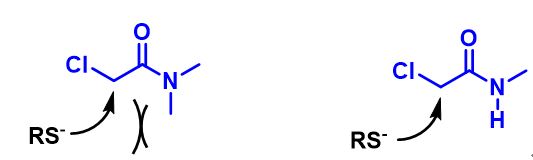
References:
[1] W. Hehre and S. Ohlinger, A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc., 2003.
[2] Spartan '20 Tutorial and User's Guide. Irvine, CA, USA: Wavefunction, Inc. 2021; p256.
[3] D.C. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems, Wiley, New York (2001).
[4] Changes in Cl-C-C-O dihedral angle of the incoming N-methyl chloroacetamide
This article is written and edited by Guijun Liu, Qiuyue Wang, Zhong Zheng, Shouliang Wang, Yongsheng Chen, John S. Wai