












网站维护
系统内容更新/升级中


在第九章《利用活化能计算预测吡唑氮烷基化的区域选择性》中,我们学习了应用活化能来判断反应的选择性以及利用活化能差来预测产物的比例的方法,并在后续多个章节用来助力反应结果预判。本章节我们进一步探讨反应底物因素对反应活化能以及区域选择性的影响,利用活化能计算在预测吡唑氮烷基化区域选择性方向的进阶考察。
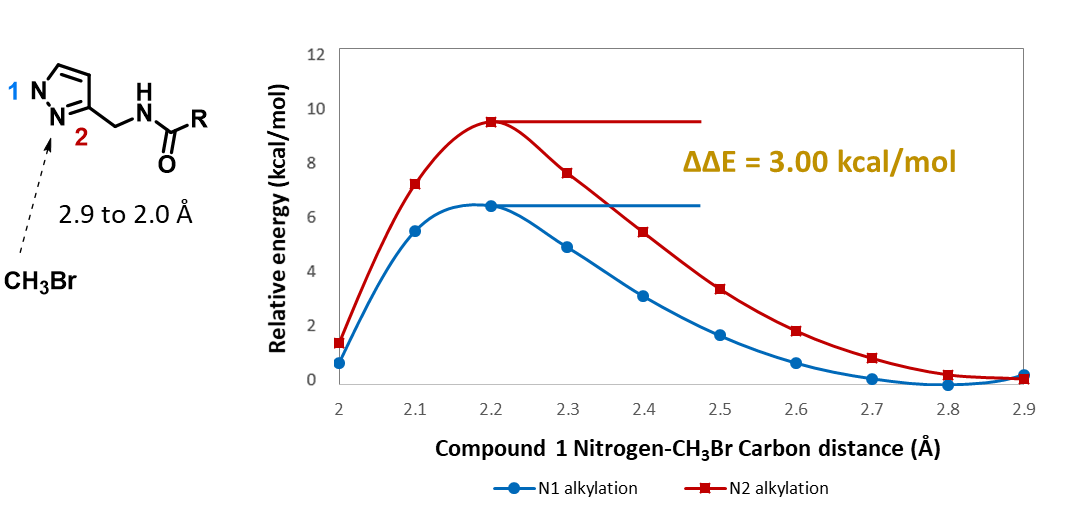
日常工作中,为了节省时间及资源,我们常采用简化底物结构的方法来做反应的初步预测。例如吡唑的烷基化反应(图1),我们通常会用CH3Br做为简化的烷基化试剂来建立反应模型,分别计算N1,N2位烷基化所需的活化能。计算数据表明,N1位发生烷基化反应生成3A所需活化能为6.4 kcal/mol,N2位发生烷基化反应生成3B所需活化能为9.4 kcal/mol,N2,N1位发生烷基化反应的活化能差高达3.0 kcal/mol,N1位更容易发生烷基化反应,产物3A将占明显优势(图2)。
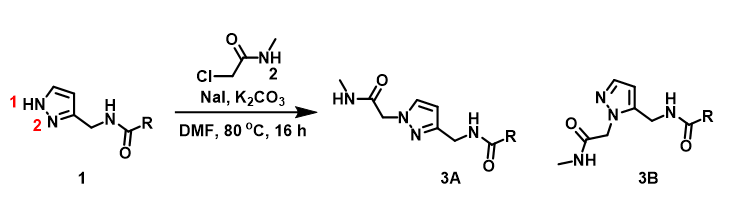
实际操作中,实验人员用氯乙酰胺作为烷基化试剂,与CH3Br有一定的差别,只有N2的烷基化产物3B生成,与计算结果完全相反。那么是什么因素导致这一结果呢?我们决定用实际的烷基化试剂结构重新计算N1,N2位发生烷基化反应的活化能,一探究竟!
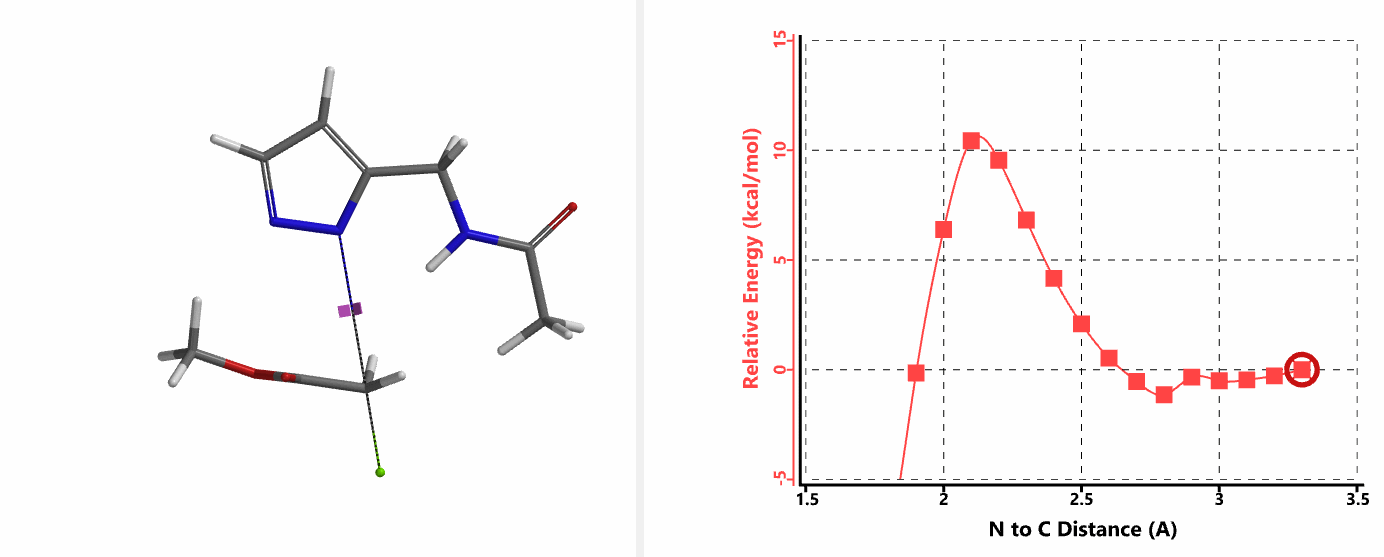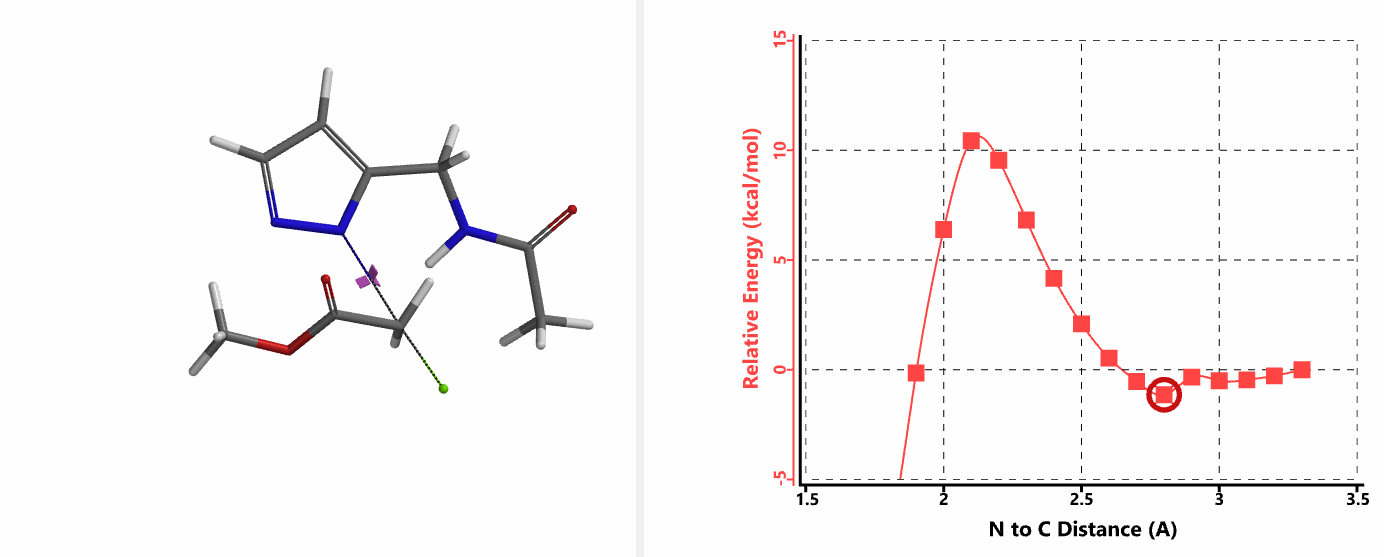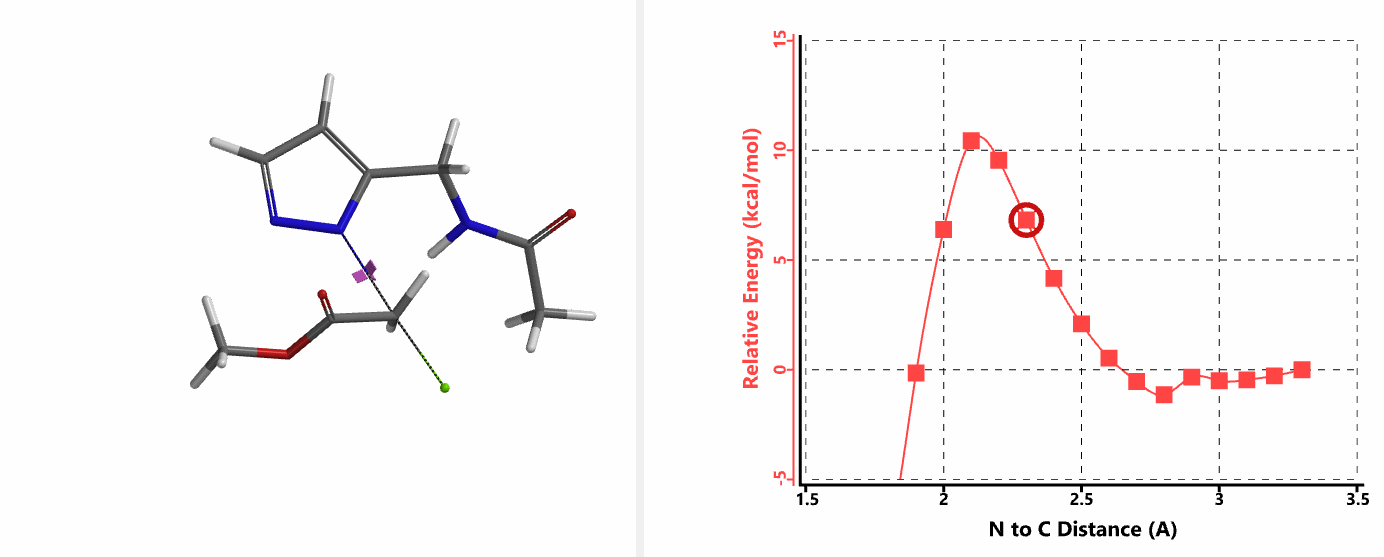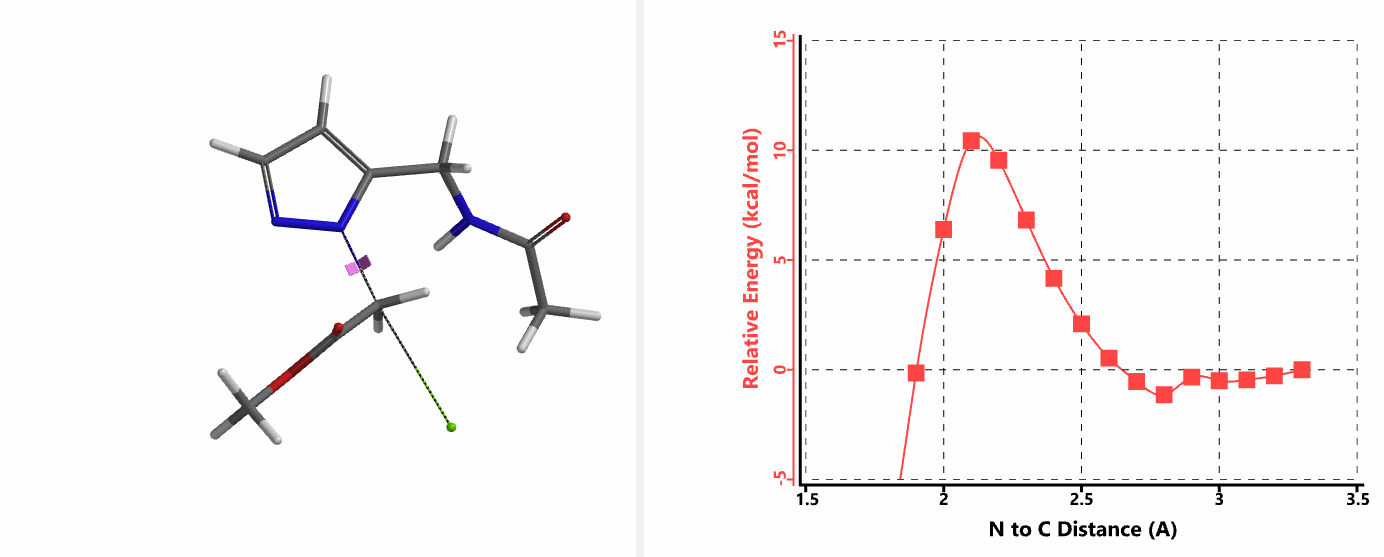
重新建立反应模型,以氯乙酰胺作为烷基化试剂,计算得到N1、N2位发生烷基化的能量曲线图。如图3所示,N1位反应所需的活化能为18.0 kcal/mol,N2位反应为15.0 kcal/mol。N1,N2位发生烷基化反应的活化能差3.0 kcal/mol。利用阿伦尼乌斯公式,可以计算得到80 ℃时,N2位产物与N1位产物的比例约为70:1,与实验结果完全匹配。
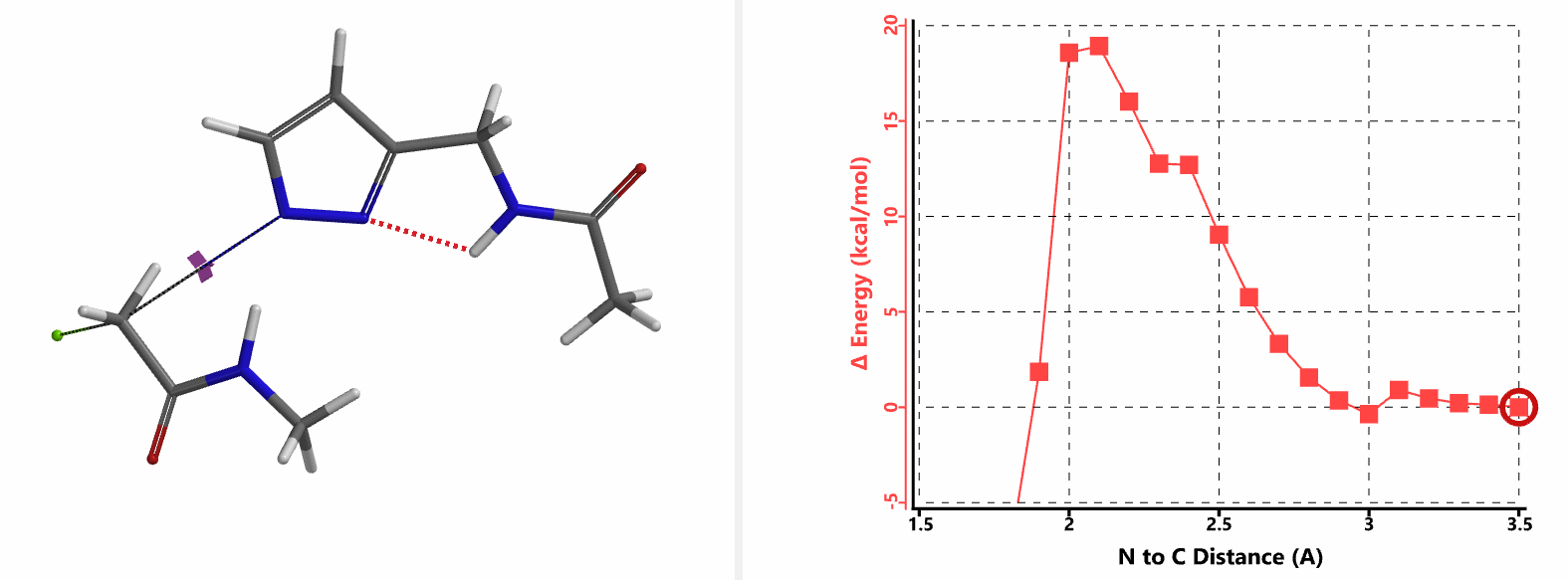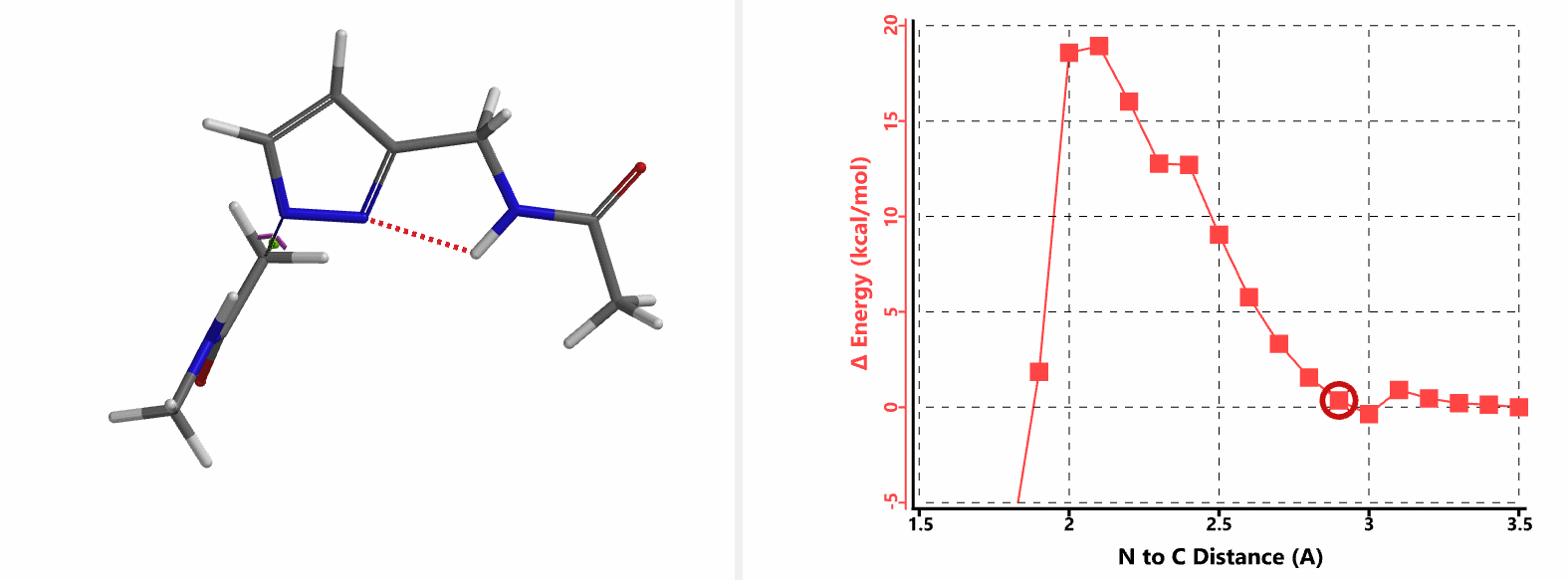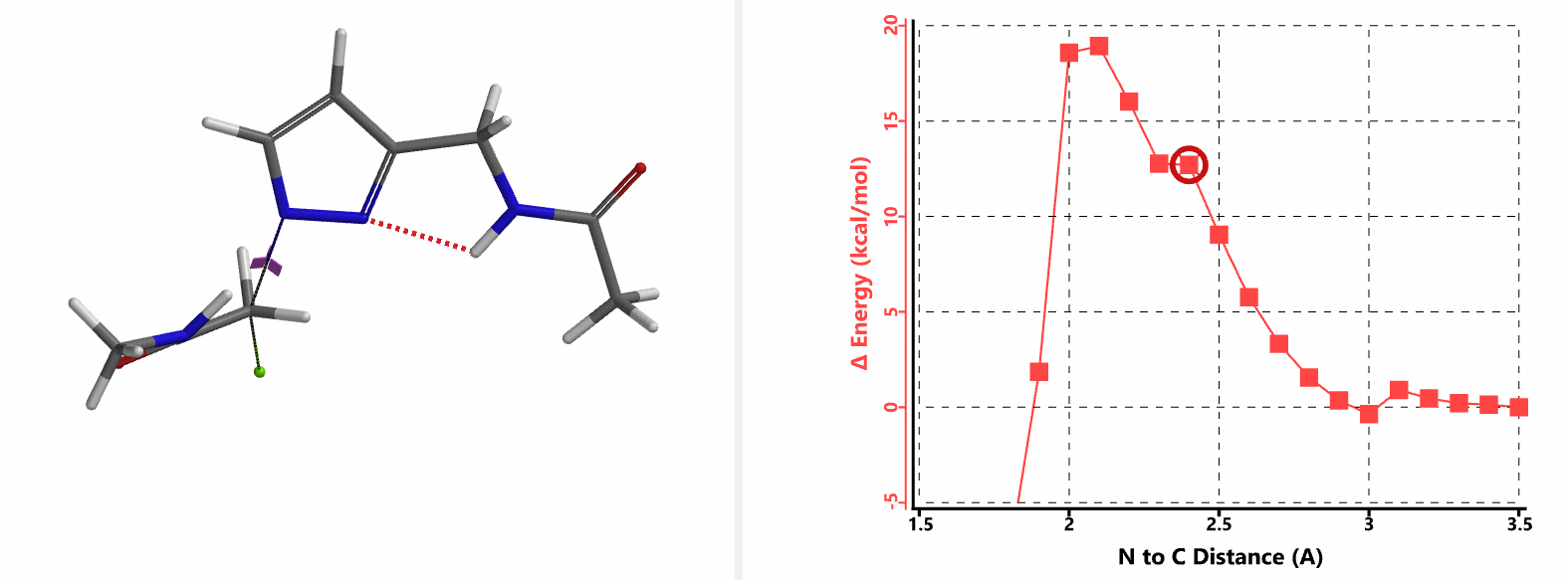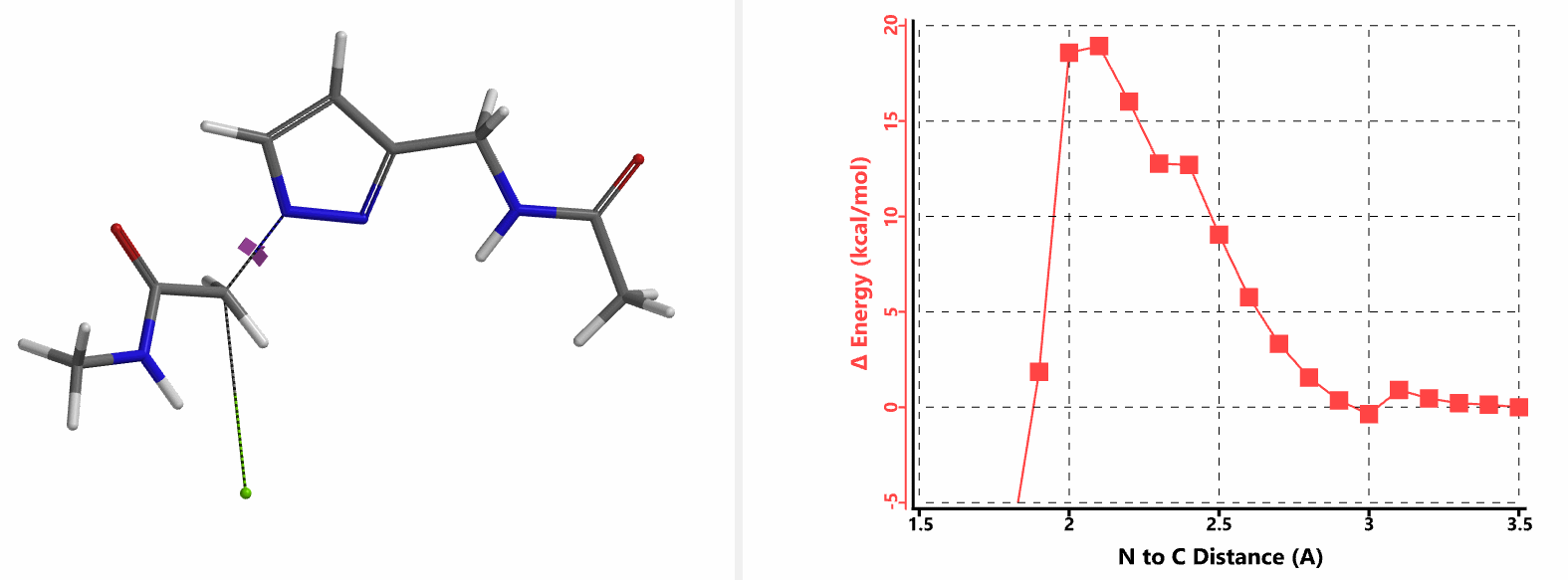
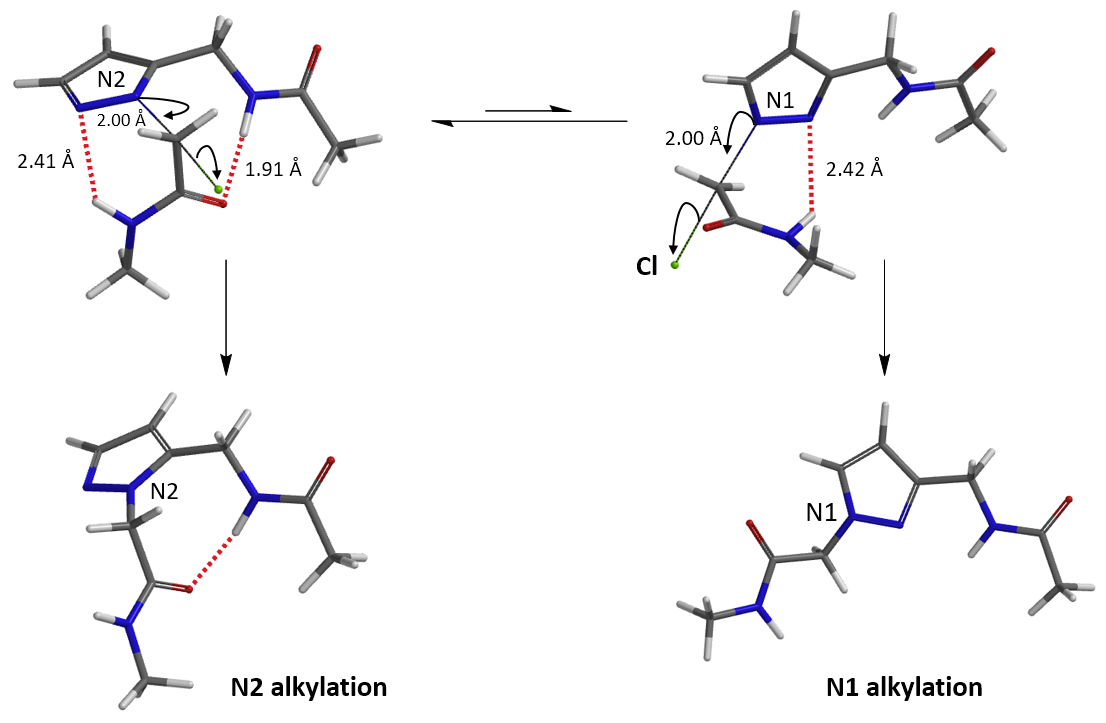
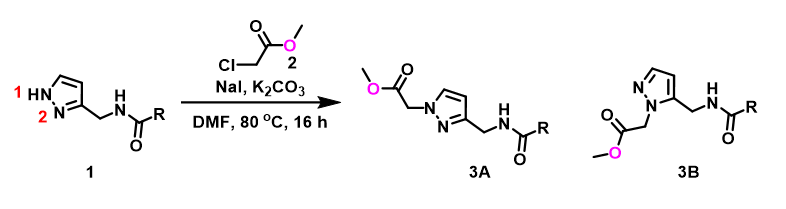
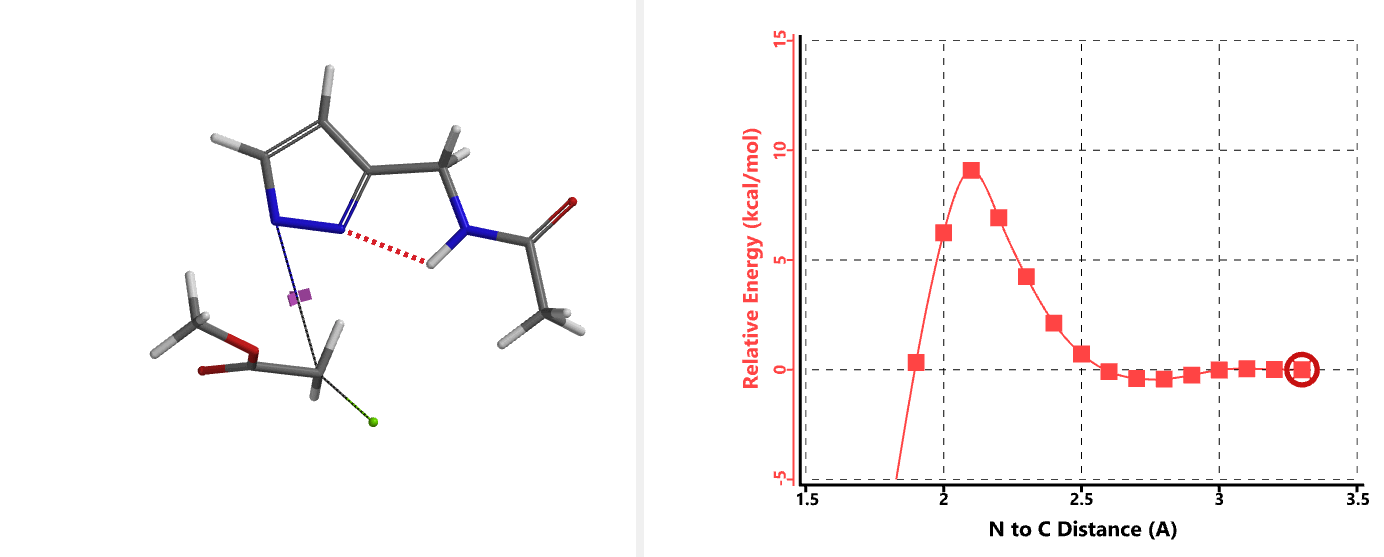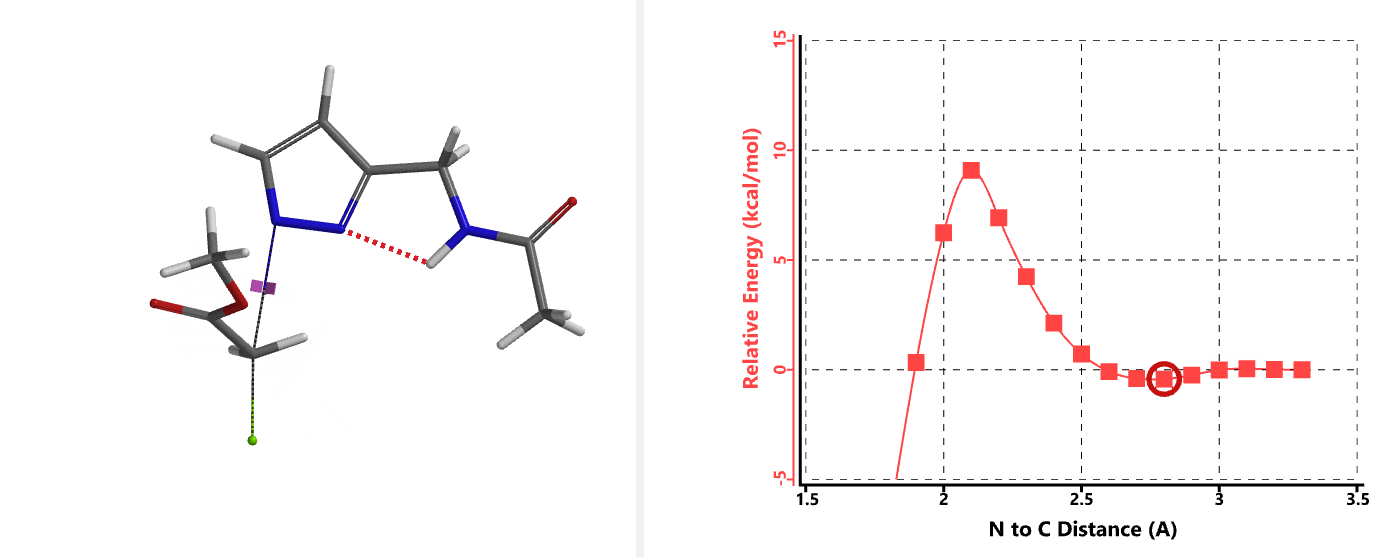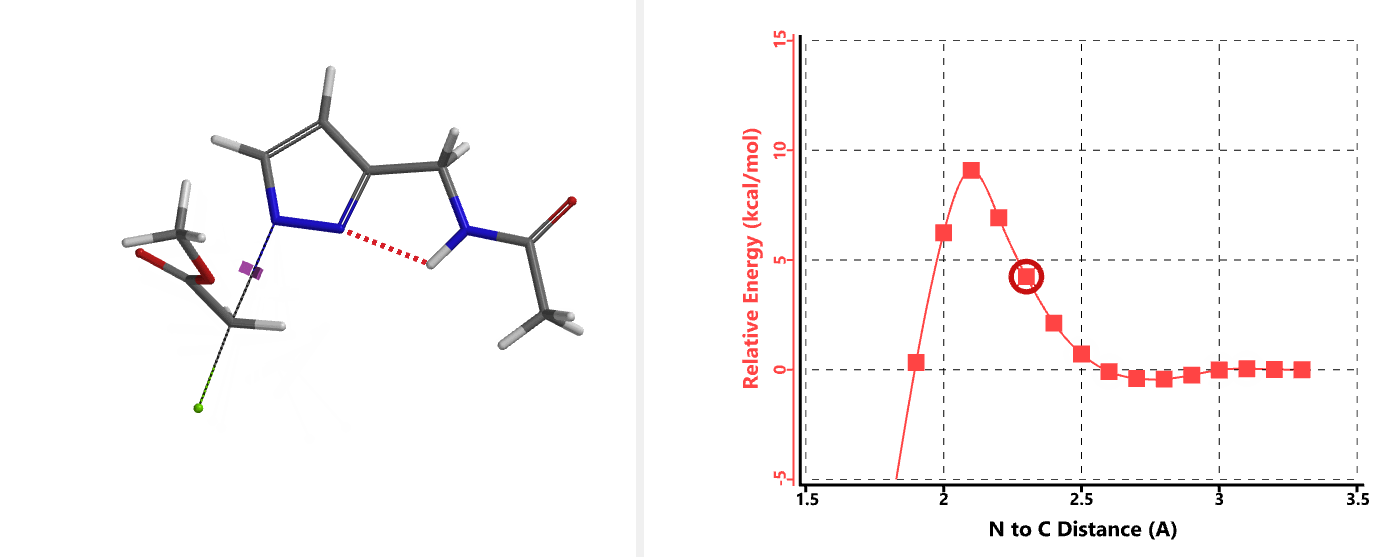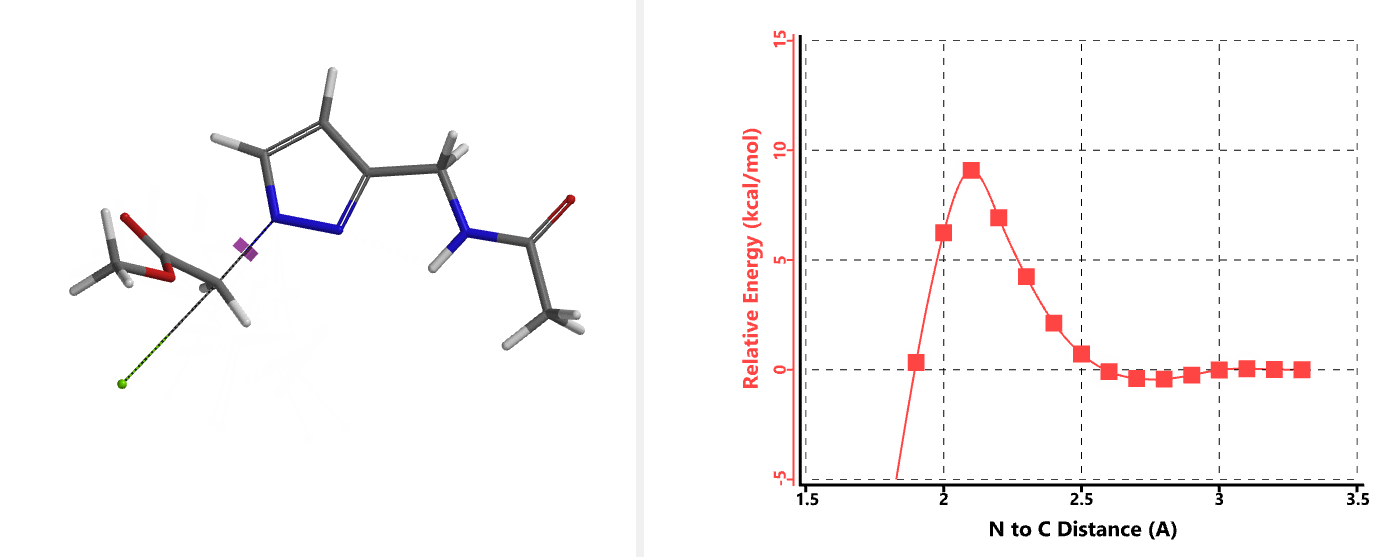
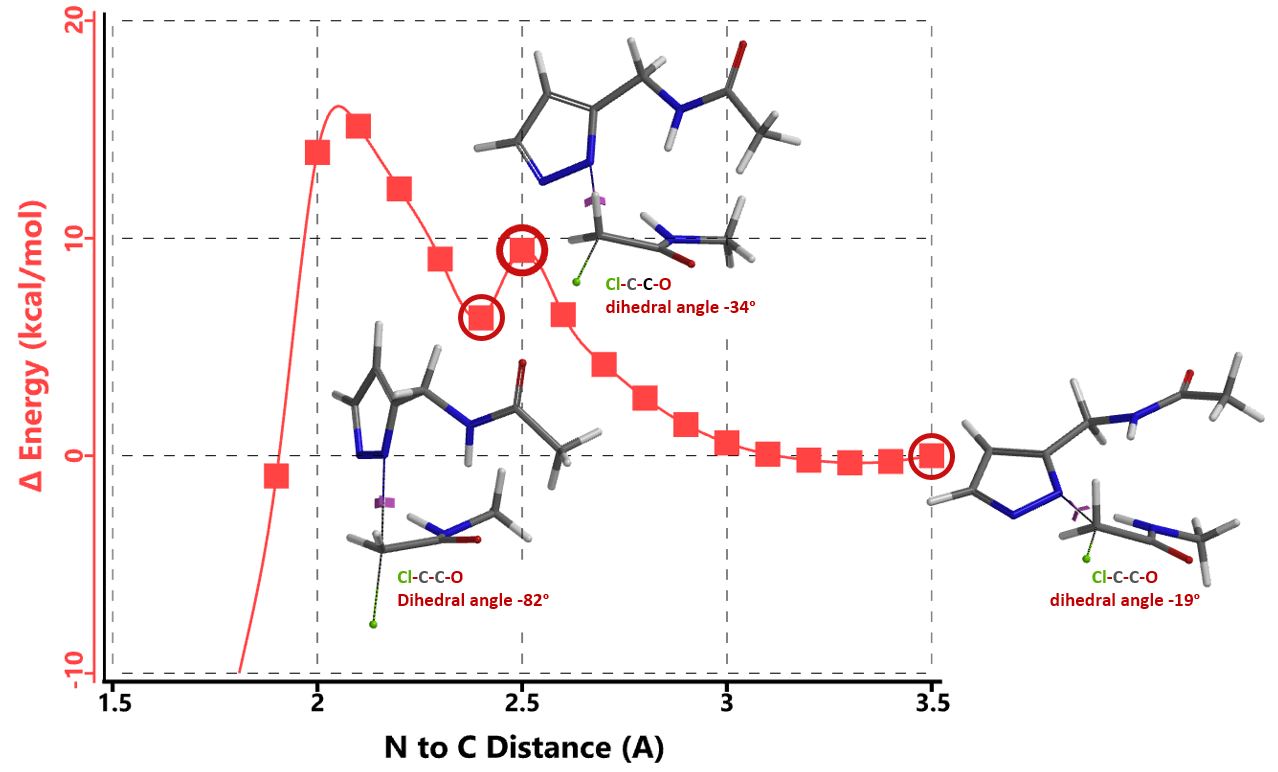
我们在N1烷基化能量曲线图中观察到一个明显的驼峰,此时可以看到N1原子从与酰胺键共平面转变为垂直方向,过渡态结构中酰胺的N-H与吡唑N2形成氢键(2.42 Å)[2],反应所需活化能值为18 kcal/mol;再来看N2位置发生烷基化反应得到的能量曲线图,过渡态结构可以观察到过渡态结构中酰胺的N-H与吡唑N1形成氢键(2.41 Å), 同时在酰胺羰基氧原子与邻位取代基NH之间形成另一个氢键(1.91 Å),反应所需活化能值为15 kcal/mol[3]。两个氢键的共同作用,使N2位发生烷基化反应所需能量远低于N1位反应所需,跨越常规影响比重大的位阻因素,得到出乎意料的高选择性单一产物[4]。
通过进一步计算过渡态(图5),发现其有且只有一个虚频,进一步验证了我们对烷基化过程的推断,肯定过渡态合理性。
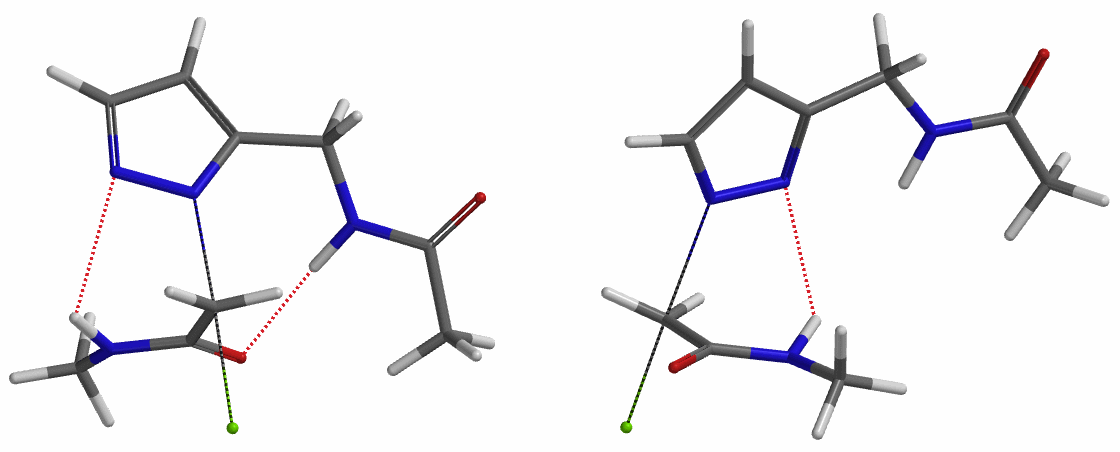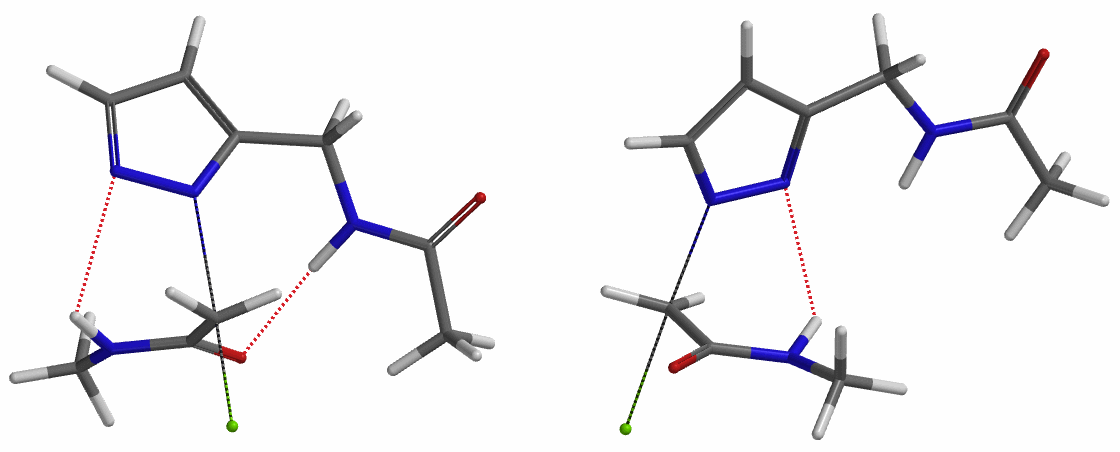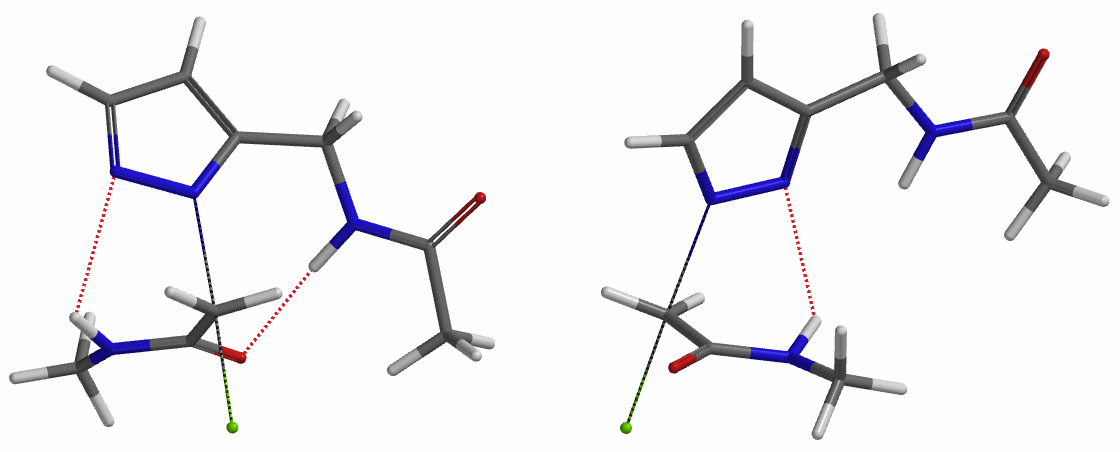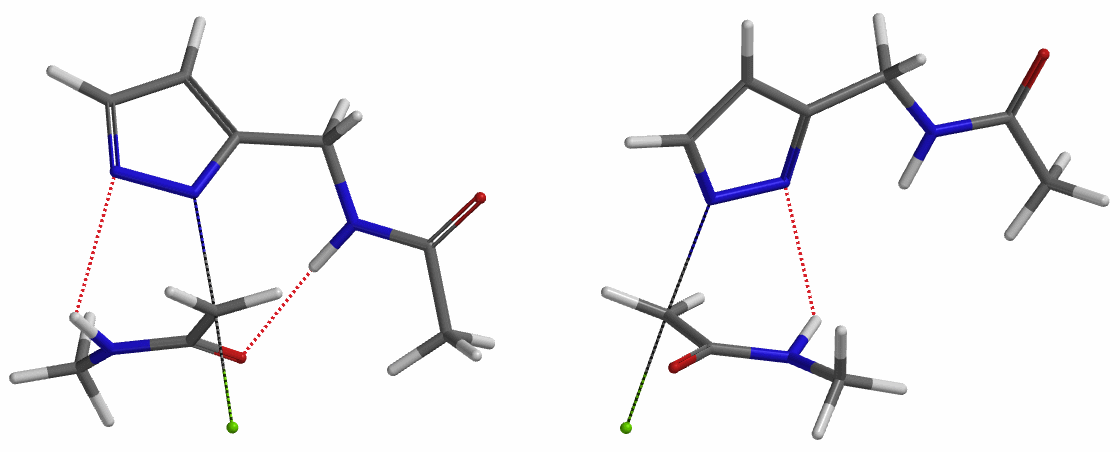
为了进一步论证分子中酰胺键NH和吡唑N形成的氢键对烷基化选择性的影响,接下来我们以氯乙酸甲酯作为烷基化试剂,排除氢键影响,考察该底物烷基化反应。如图6所示,N1烷基化所需的活化能为9.5 kcal/mol,N2烷基化为11.6 kcal/mol,N1烷基化所需的能量更低,反应更容易发生在这个位置,这与实验结果也相吻合。
这个案例向大家展示了QM计算过程中模型构建的重要性。计算模型的构建往往需要综合考虑多个因素,比如底物结构的简化替代实际底物计算是否合理,底物和烷基化试剂产生的分子间作用力,溶剂体系参与带来的影响(第24章《应用QM研究重氮转移反应的机理》)等等。理论可以指导我们的实践,反过来实践积累的经验可以增强我们对理论的理解与运用。希望这个例子能让大家得到启发,使QM能更好的运用到自己的工作中。
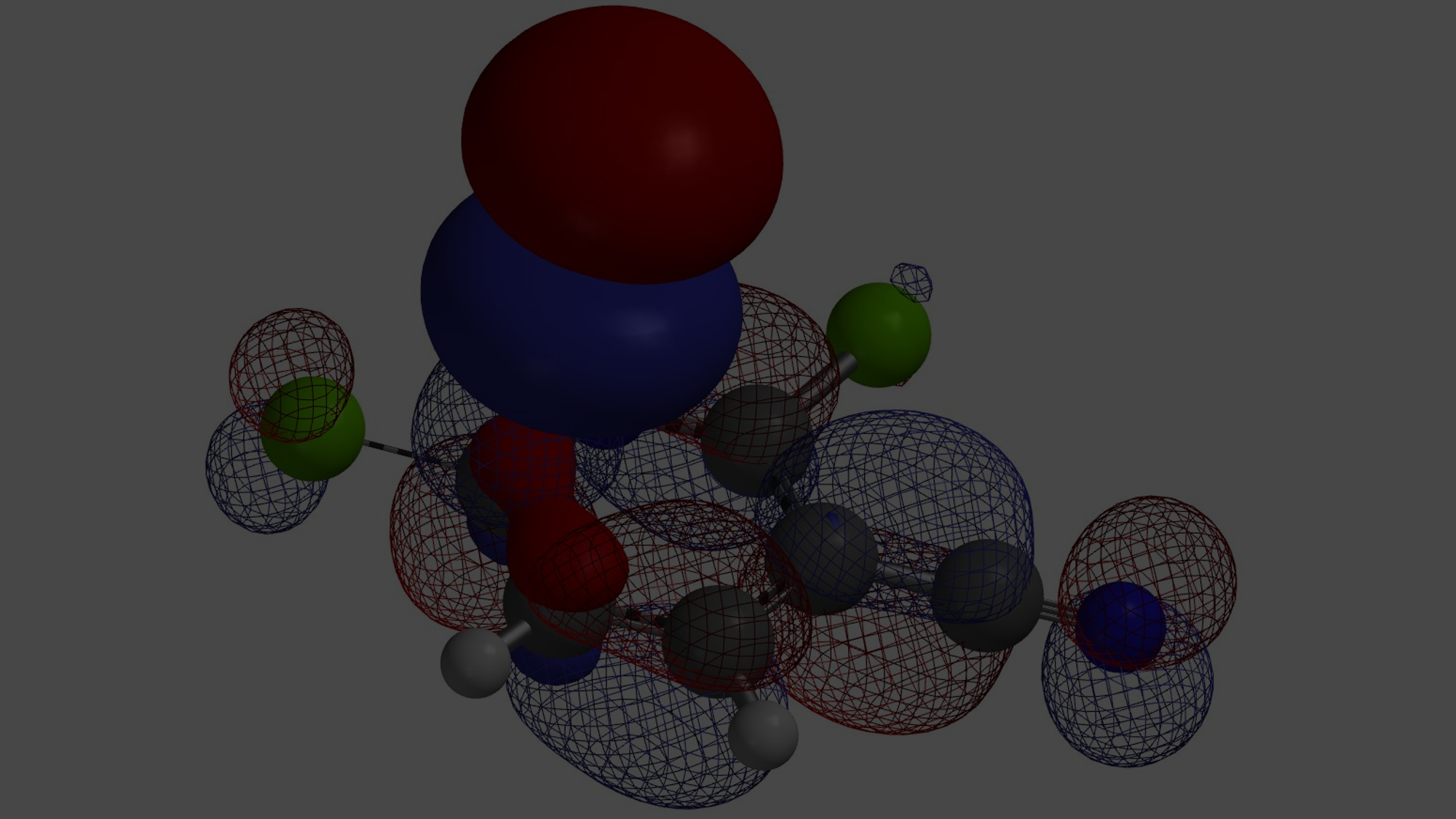
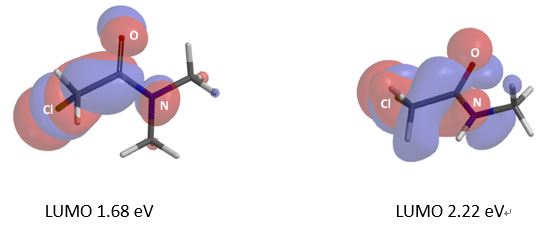
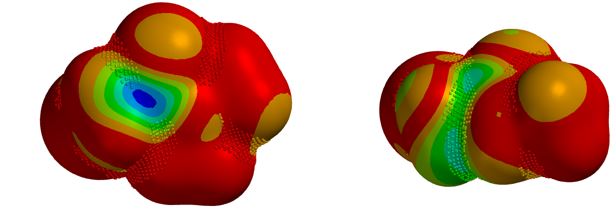
在正文部分,我们学习到氢键在反应过程中起到了关键的作用。由于氢键作用力的参与,反应得到异于经验的位置异构产物。现在我们思考下面这个例子,硫醇作为亲核试剂,N,N-二甲基氯乙酰胺、N-单甲基氯乙酰胺为底物发生亲核取代反应,在常规条件下,哪个反应速度更快呢?计算结果表明二者LUMO map有明显差异(图8),而活化能计算曲线表明氢键在这种情况下会导致反应减慢,大家可能已经想到其中缘由。期待你的留言,神秘大奖等着你!
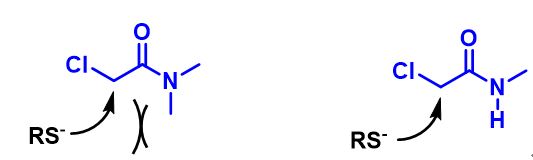
参考文献:
[1] W. Hehre and S. Ohlinger, A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc., 2003.
[2] Spartan’20 Tutorial and User’s Guide. Irvine, CA, USA: Wavefunction, Inc. 2021; p256.
[3] D.C. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems, Wiley, New York (2001).
本文由刘贵军、王秋月、郑重、王守亮、卫小文编撰。
