












网站维护
系统内容更新/升级中


Over the years, we received a lot of questions from our readers, below are the frequently asked ones.
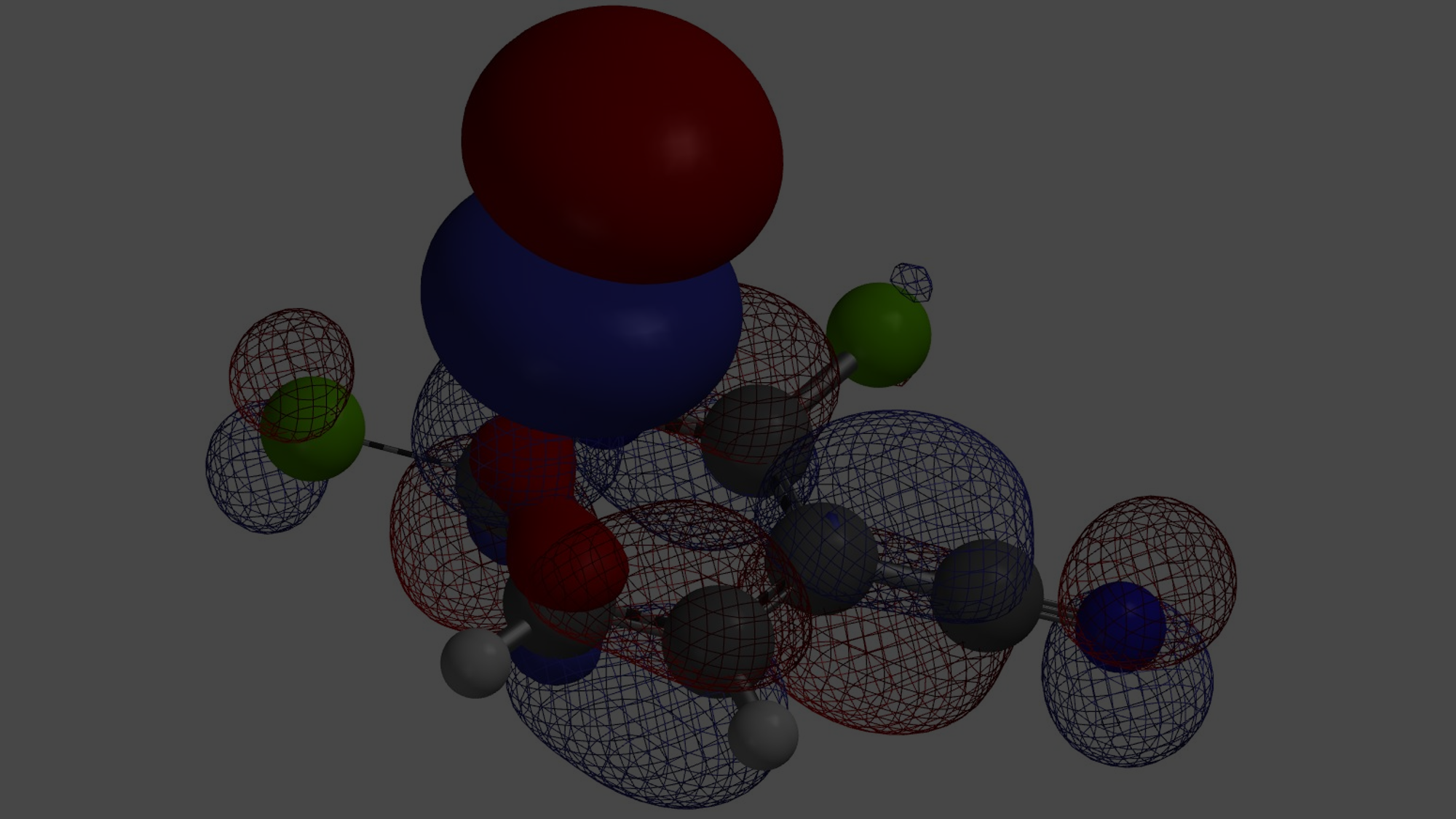
Gaussian, Gamess, and ORCA are popular ones for organic chemists. WuXi AppTec provides us with Spartan, which uses Q-Chem as the calculation engine.
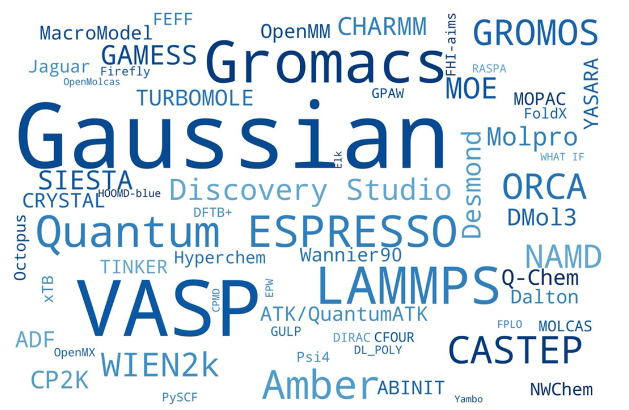
Currently we use Density Functional Theory wB97X-D and basis set 6-31G* as our standard method. It provides a balance of speed and accuracy our organic chemists need.

We searched for practical QM textbooks which could be useful for organic chemists. There is none. As such, our colleagues felt the need to share what they learned in articles published in https://chemistry.wuxiapptec.com/qm and https://www.wuxibiology.com/magical-power-of-qm. They are collated into eBook you could download for free from the same links.

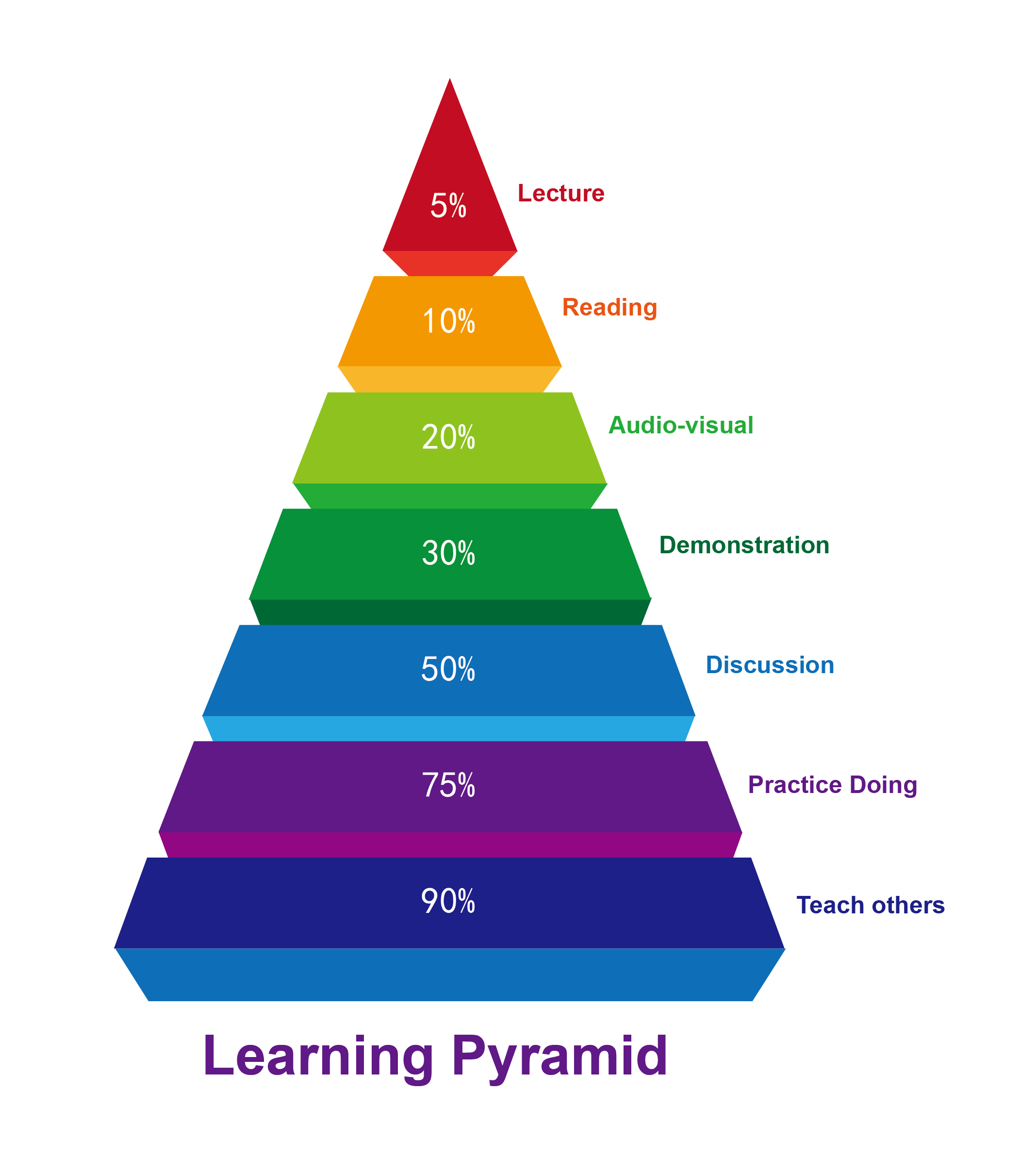
We started from retrospective analyses of reactions we run every day, especially the challenging ones we couldn’t predict properly intuitively. We optimized our methods, developed SOP, and turned QM into a prospective tool. We discussed and taught one another what we learned. It is a continuous team effort.
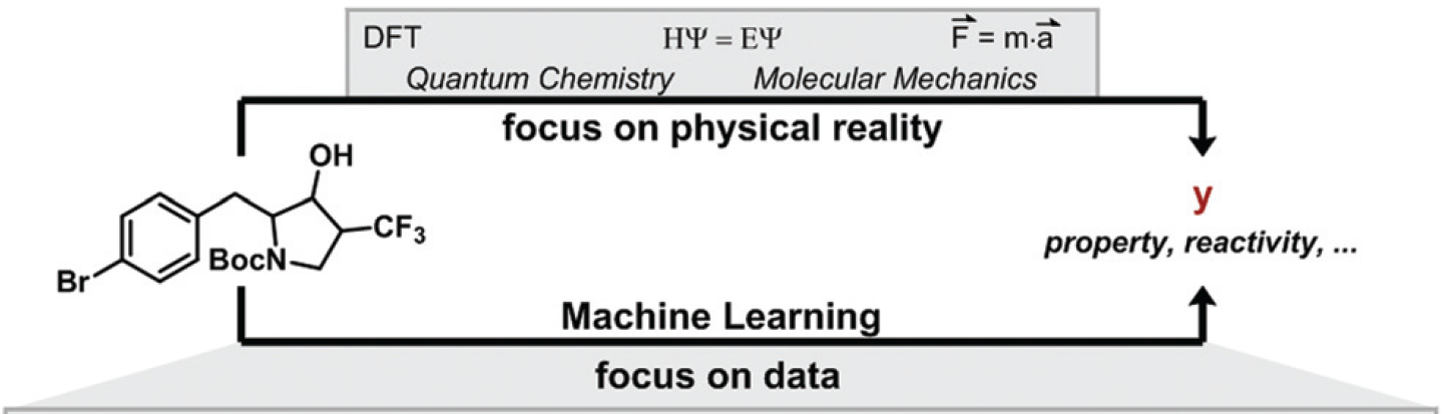
QM focuses on physical reality, could pin point uniqueness of each substrate, each reagent. ML focuses on data, statistical average. Currently QM offers better guidance when it is not intuitive easy to make correct prospective analyses and is more useful in retrospective analyses for divergence in reactivity. As QM parameters are incorporated into Machine Learning, we anticipate chemistry ML tools to continue to improve.
Literature calculations that are useful for practical analyses are very limited. It is a continuous efforts in WuXi AppTec to develop these methods. We delegate our QM analyses to chemists with appropriate levels of proficiency and encourage everyone to learn. As we invest our efforts to analyze reactions of interest, we become more proficient. QM calculation methods and computational power are evolving very fast, new reactions continue to be discovered, etc. Over time, we learn “What to calculate, How to calculate, How to interpret the results, and How to improve the methods.” This is lifelong learning.

We established SOP for different analyses, fit for purpose ones. From orbital to electrostatic potential map analyses which take a few minutes, to reaction energy profiles, transition state calculations which could take a few hours. Accessible QM Databases with properties of a large number of organic molecules pre-calculated are tremendously useful.

QM calculations are CPU intensive, good to run them on desktops with ≥8 cores processors, which have become more affordable. The balance between computational vs time+labor+ reagent+disposal costs has changed significantly. QM calculation is becoming a valuable Resource Optimization/Green Chemistry tool.

For specific computational chemistry needs, please email IDSU_Operation@wuxiapptec.com
This article is written and edited by Yongsheng Chen and John S. Wai.
References:
[1] L. Talirz, L.M. Ghiringhelli, B. Smit, Living Journal of Computational Molecular Science, 2021, 3(1), 1483.
[2] a) R. Peverati, Int. J. Quantum Chem. 2021;121:e26379 b) L. Goerigk, N. Mehta, Aust. J. Chem. 2019, 72, 563.
[3] a) F. Strieth-Kalthoff, F. Sandfort, M.H.S. Segler, F. Glorius, Chem. Soc. Rev., 2020, 49, 6154. b) Y.F. Guan, C.W. Coley, H.Y. Wu, A. Ranasunghe, E. Heid, T.J. Struble, L. Pattanaik, W.H. Green, K.F. Jensen, Chem. Sci., 2021, 12, 2198.
[4] https://www.davidparkins.com
[5] a) Spartan Spectra and Physical Properties Database: https://www.wavefun.com b) QM7-X: J. Hoja, L.M. Sandonas, B.G. Ernst, A. Vazquez-Mayagoitia, R.A. DiStasio Jr, A. Tkatchenko, Sci Data, 2021, 8, 43.