












网站维护
系统内容更新/升级中


Electrophilic aromatic substitution (SEAr) is a very important class of reactions for chemists to introduce functional groups onto heterocycles.
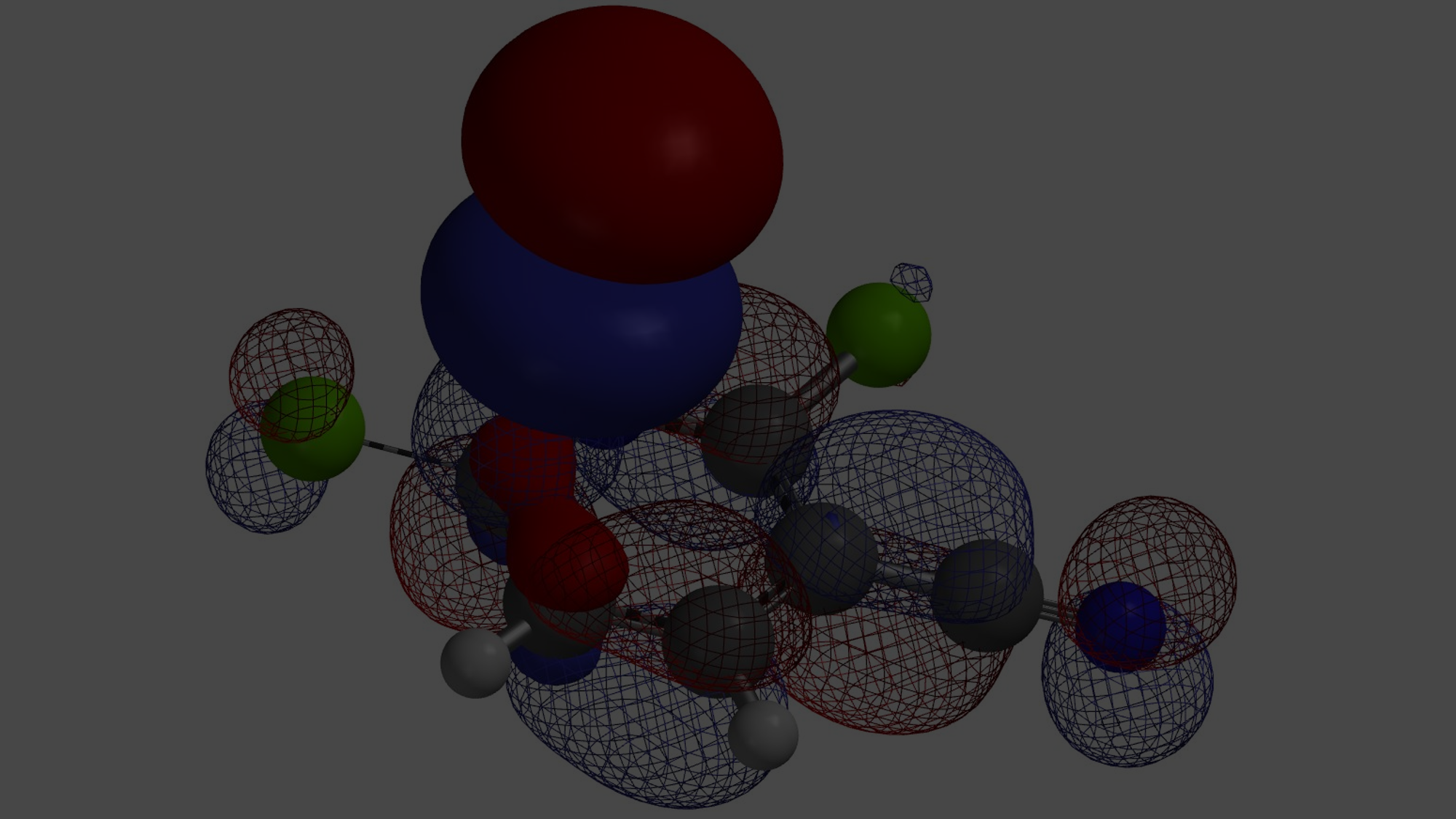
We all learned that substituents on aromatic rings affect regioselectivity of electrophilic substitutions. With mono-substituted phenyl analogs, if the substituent is an electron-donating group, such as a methoxy group, we usually observe reactions at ortho- and para-positions. On the other hand, when the substituent is an electron-withdrawing group, such as a nitro group, meta-substituted products are usually produced. Halogen substituents are unique, they are electron-withdrawing, yet ortho- and para-directing (Figure 1).

We also learned that predicting regioselectivity of SEAr on complex multi-substituted heterocyclic systems could be more challenging.
For instance, with substrate A (Figure 2), which heterocycle would be halogenated first, the pyridazine or the pyrazole ring? For substrate B, would halogenation be on the fused ring or on the benzene ring, and at which position? For substrate C, it is quite clear that the imidazole would be halogenated. Will we have selective halogenation at C-2 or C-3, or a mixture of products? Intuitively, these are not easy to predict with high confidence.
In this article, we will discuss how we use calculated HOMO and 13C NMR data to make predictions on the regioselectivity of EAS on heterocyclic aromatic systems.
Frontier Molecular Orbital (FMO) Theory emphasizes the use of Highest Occupied Molecular Orbital (HOMO) for analyses of nucleophiles and Lowest Unoccupied Molecular Orbital (LUMO) for electrophiles.
In Chapter 1, we showed how we utilize LUMO and LUMO Map to analyze nucleophilic aromatic substitutions. Here we will be looking at HOMO of three heterocyclic substrates (A, B, & C, Figure 3) to rationalize the regioselectivity observed with SEAr.
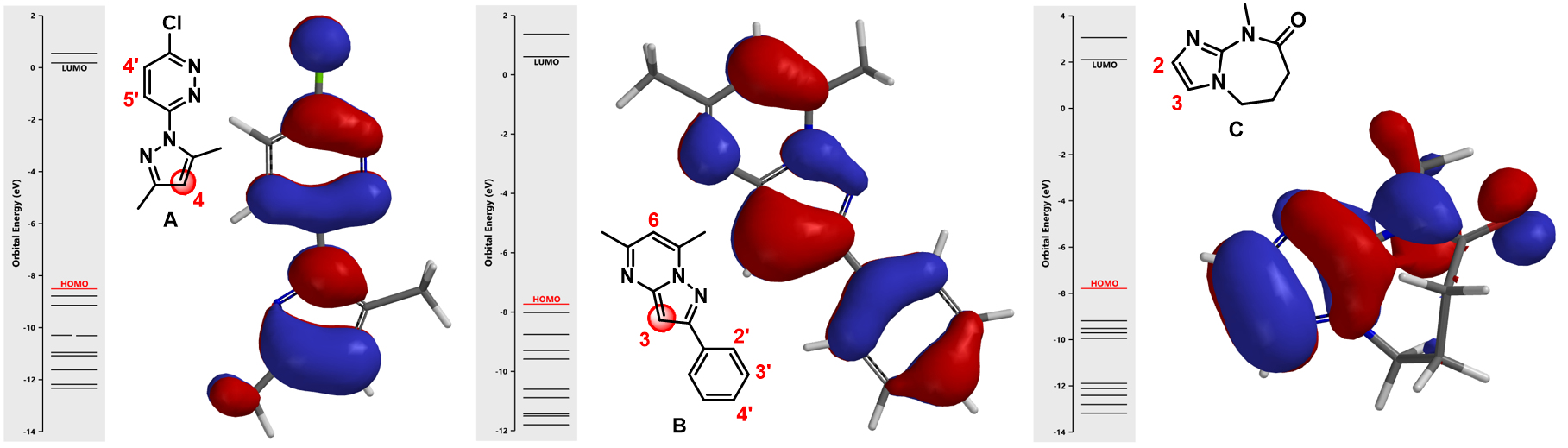
For substrate A, among three potential sites, there are HOMO lobes on C-5’ and C-4, which suggests only these two positions are capable of interacting with incoming electrophiles. The HOMO lobe over C-4 is much larger than the one over C-5’ indicating that electrophiles would react selectively at C-4. Indeed we observed only the C-4 halogenated product experimentally. Similar for substrate B, the C-3 carbon on the fused ring is associated with the largest HOMO lobe among all potential sites of reaction, resulting in selective halogenation at C-3.
Things get slightly complicated for substrate C. The HOMO lobes cover C-2 and C-3 are similar in sizes on both of atoms. In this case, will we observe a mixture of substitution products? What else have we learned to predict the regioselectivity in such circumstances?
We found QM-calculated 13C NMR chemical shift numbers of these carbons correlate quite well with regioselectivity of SEAr.
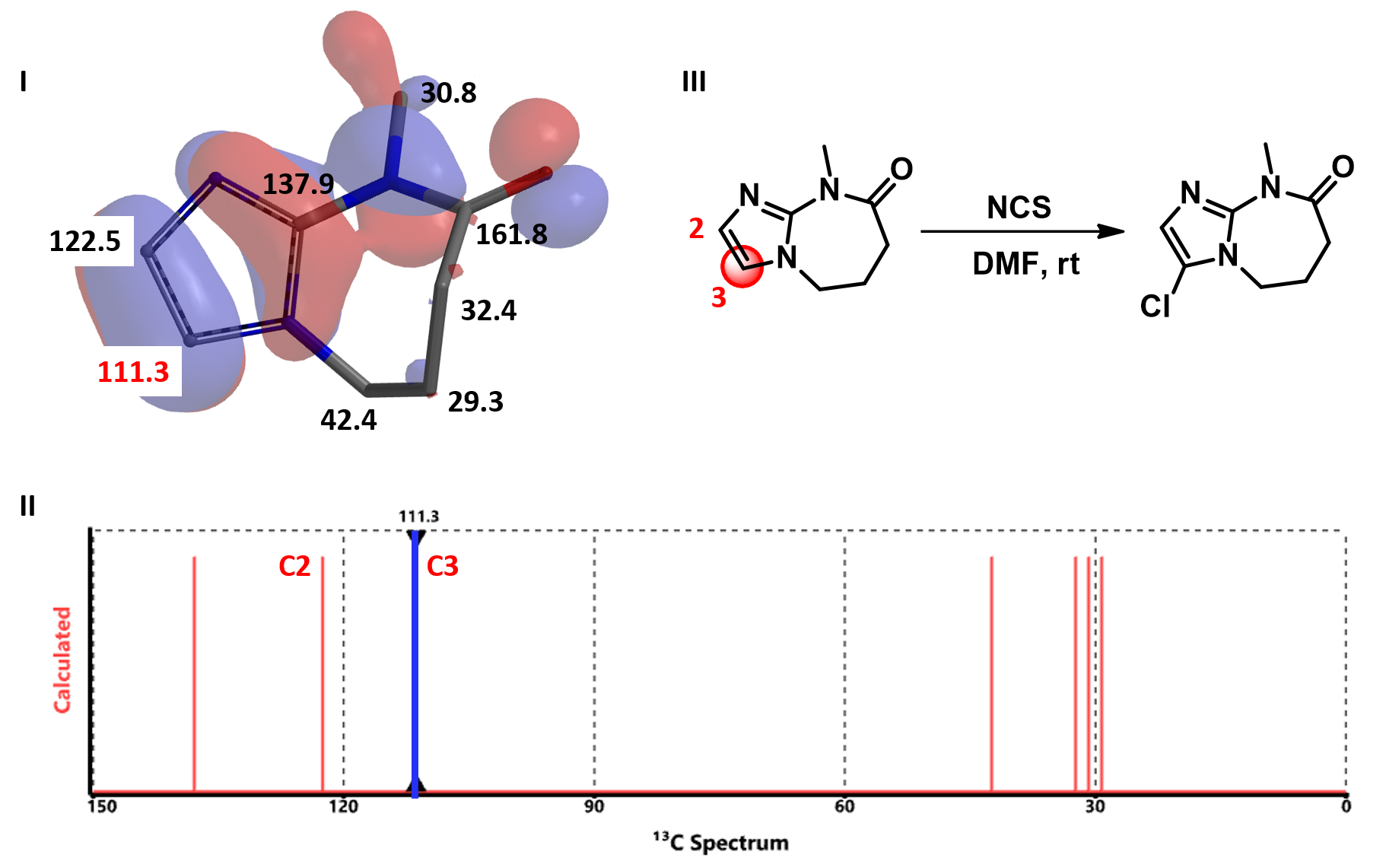
Calculated 13C NMR of substrate C (Figure 4) shows a significant difference between the chemical shift values of C-2 (122.5 ppm) versus C-3 (111.3 ppm). A lower chemical shift value on C-3 suggests a larger electron density on the carbon and C-3 halogenation to dominate. Indeed the reaction is observed to be highly selective at C-3.
In summary, by integrating QM-calculated HOMO and 13C NMR data of the substrates, we could rationalize the electrophilic reactions and then predict regioselectivity reliably. This becomes part of the standard QM evaluations we do routinely in our retrosynthetic planning.
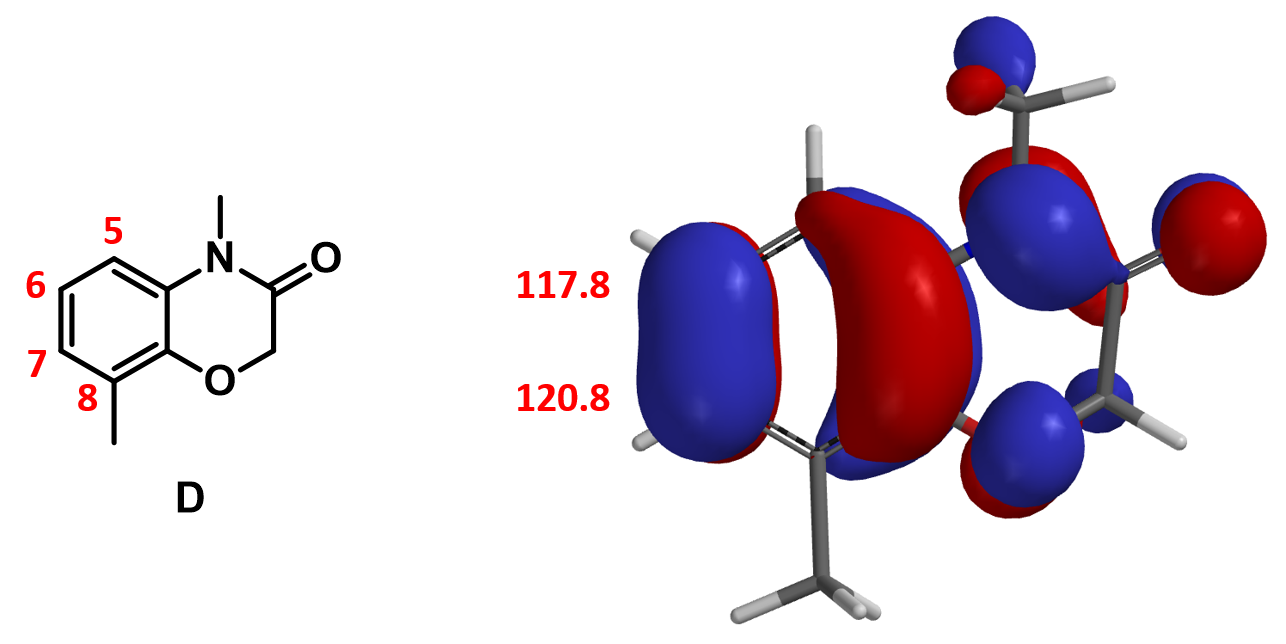
On substrate D, there are three potential sites that could be brominated. They are C-5, C-6, and C-7. Since there is no HOMO lobe associated with C-5, we only need to evaluate relative reactivity at C-6 and C-7. However, differences in HOMO lobe volumes and the chemical shift values of C-6 and C-7 are not significant, and steric effects from C-8 methyl group could play a role. In our future article, we will introduce additional QM method that could help us make reliable prediction in such situation.
In next article, we will discuss the application of electrostatic potential map to estimate pKa for selective deprotonation.
This article is written and edited by Qiuyue Wang, Guqin Shi, Jian Wang, John S. Wai.
References:
[1] Warren J. Hehre (2003). A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc.
[2] F.A. Carey & R.J. Sundberg (2007). Advanced Organic Chemistry Part A: Structure and Mechanisms. New York, NY, USA: Springer Science+Business Media, LLC. Pg.771-779
[3] M. Kruszyk, M. Jessing, J.L. Kristensen, M. Jorgensen, J. Org. Chem. 2016, 81, 5128