












网站维护
系统内容更新/升级中


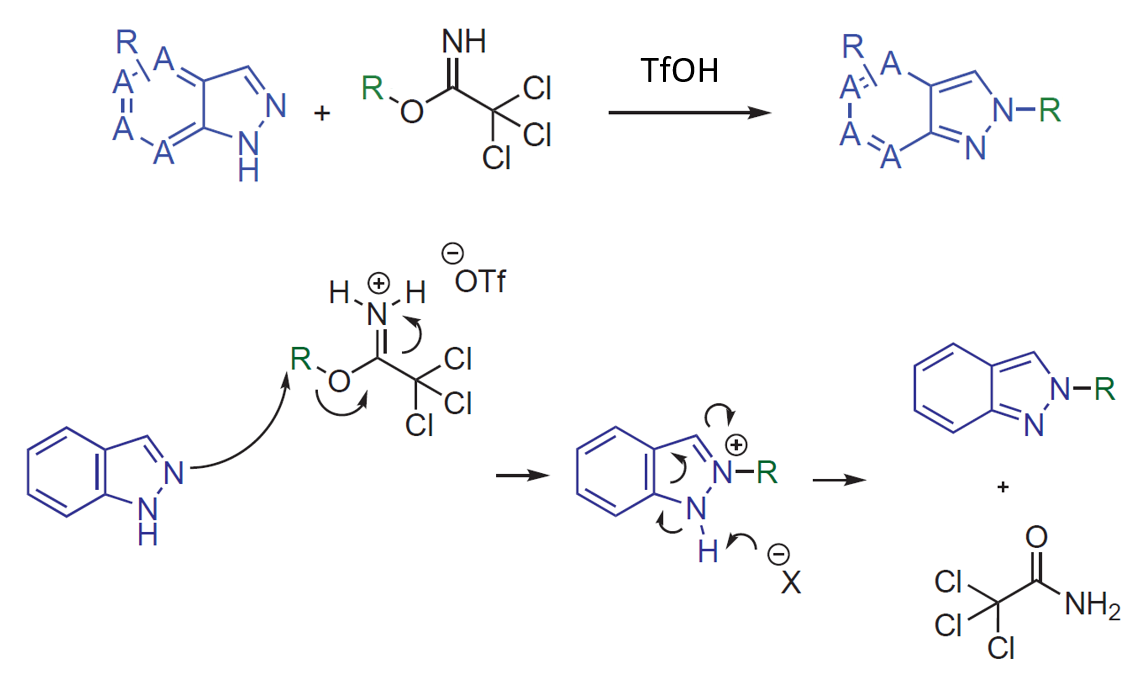
2022年辉瑞的化学家们在Synthesis杂志上发表了一篇吲唑类化合物与伯、仲和叔烷基2,2,2-三氯乙酰亚胺酯高选择性N2烷基化反应的文章[1]。文章给出的示例中,无论吲唑取代基的电性如何,都高选择性的获得了N2烷基化产物,没有观察到N1异构体。作者提出的反应机理如图1所示,在酸性条件下,三氯乙酰亚胺酯的亚胺氮原子发生质子化,被活化后与吲唑2位氮发生亲核取代反应,芳构化后生成相应的2-烷基吲唑。
今天,我们将利用QM工具来进一步验证文章中提出的机理。
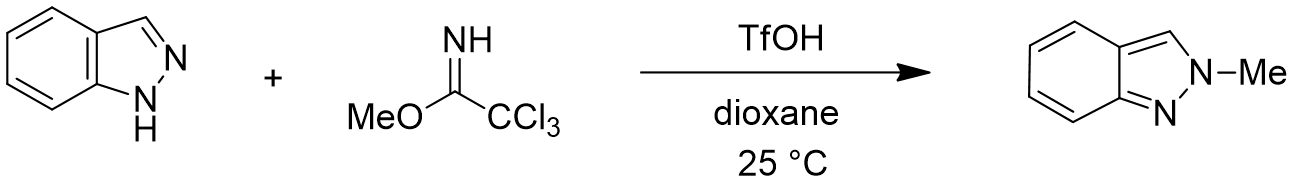
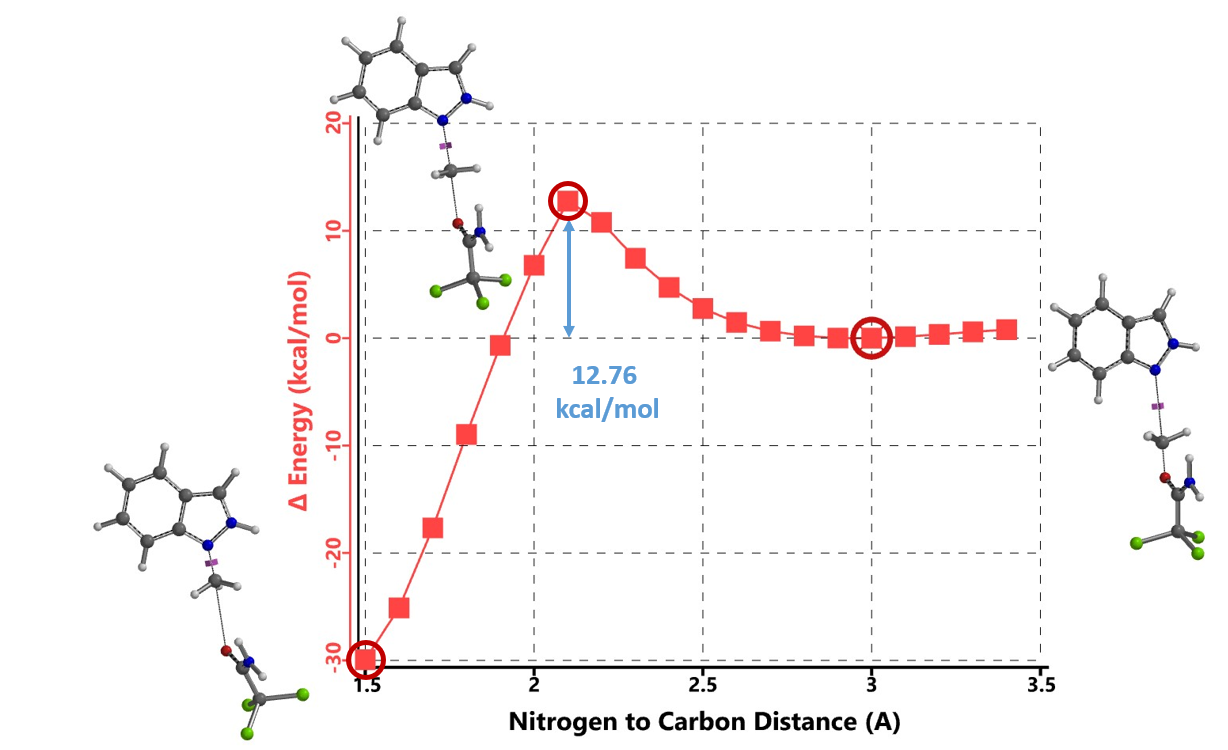
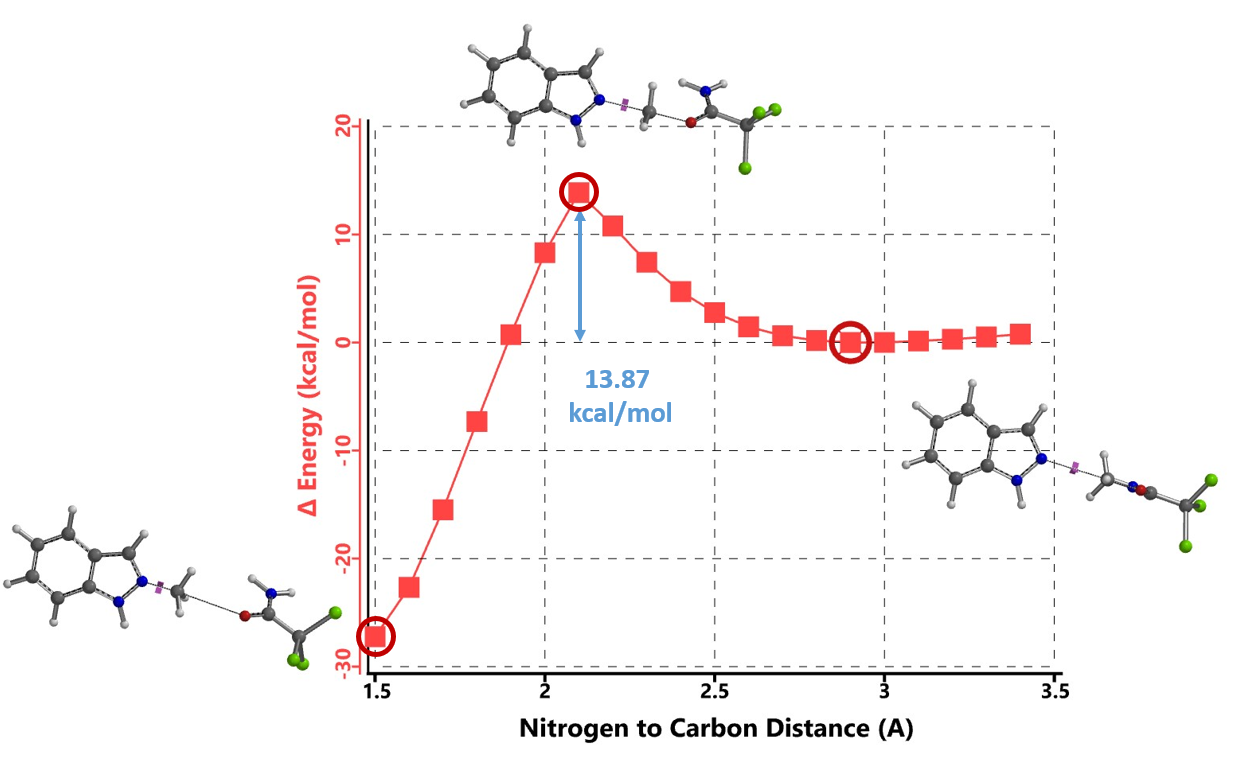
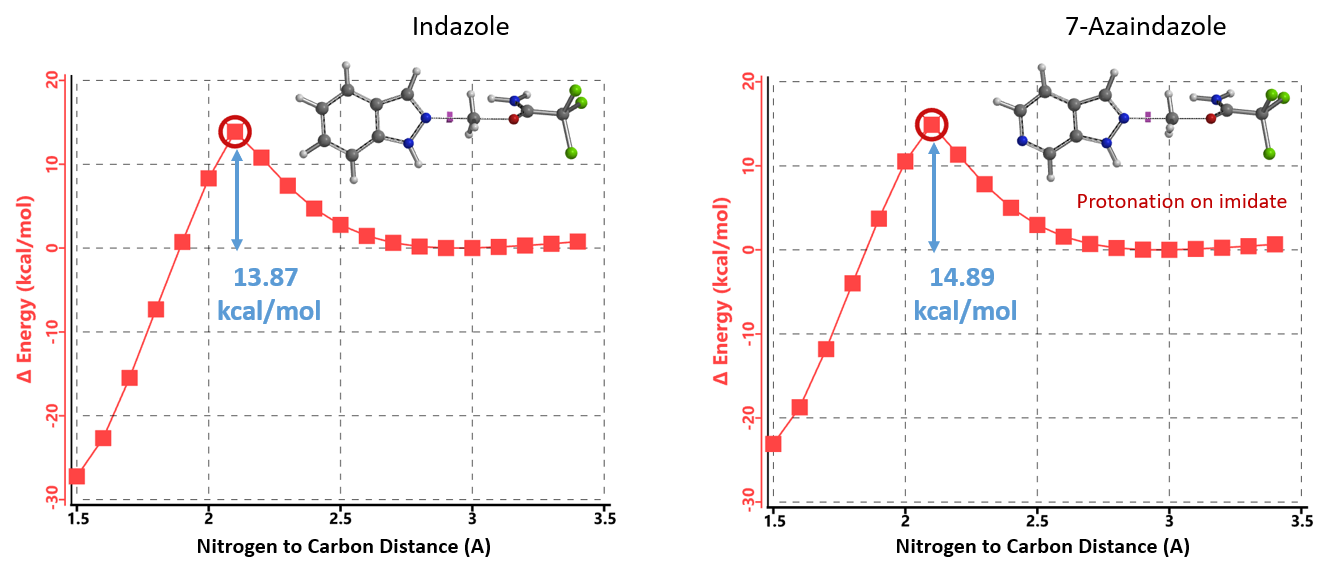
首先,我们选择了文章中结构相对简单的吲唑和三氯乙酰亚胺甲酯两种底物为模型,使用Spartan (DFT wB97X-D/6-31G*) 分别计算了N1和N2烷基化反应的能量变化曲线,发现N1烷基化所需的活化能约为12.76 kcal/mol(图2),N2为13.87 kcal/mol(图3)。N1烷基化所需活化能比N2低1.11 kcal/mol,那么是不是可以就此说N1烷基化更容易发生呢?答案是否定的,我们还需要考虑1-氢吲唑和2-氢吲唑两种互变异构体(tautomers)的相对能量。
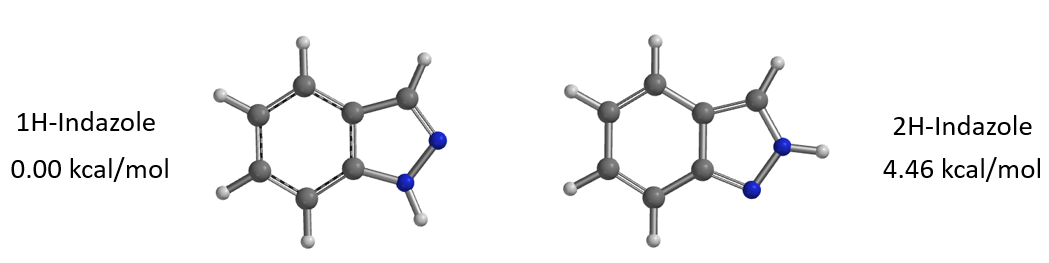
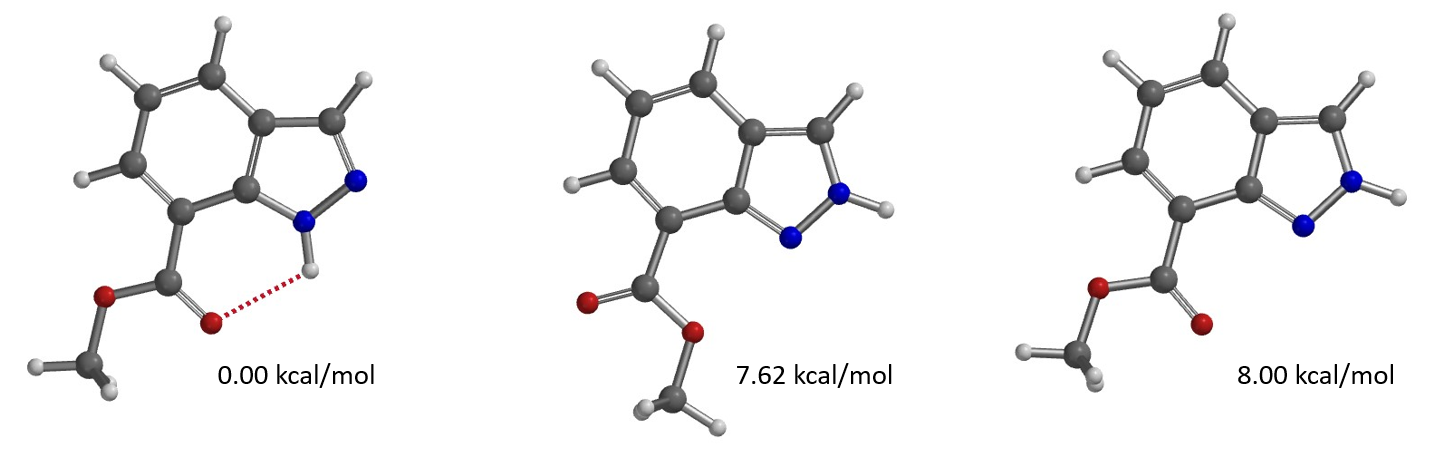
通过计算可知1-氢与2-氢吲唑互变异构体之间的能量差为4.46 kcal/mol,2-氢吲唑的能量相对较高(图4),它需要从能量较低的1-氢吲唑克服一定的能量转变而来,这一点分析时需要考虑进去。
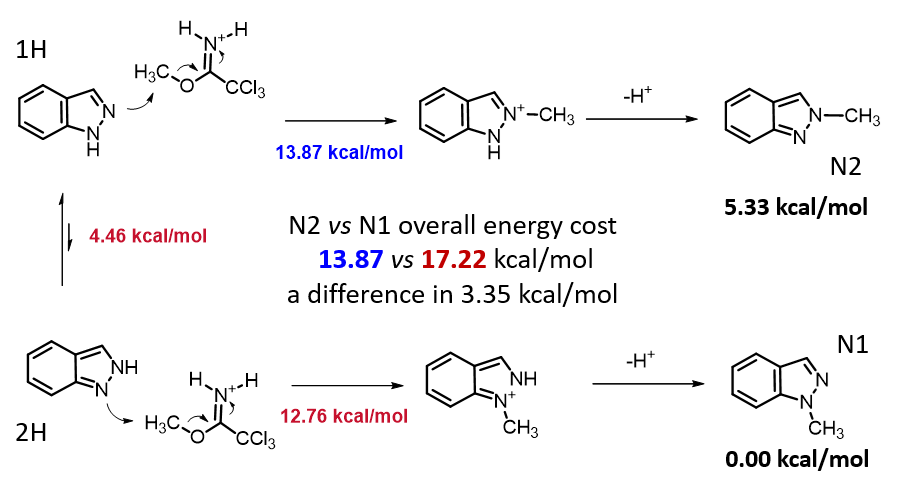
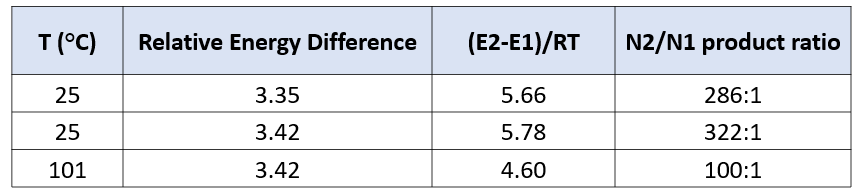
两种反应途径的实际反应历程如图5所示,N1烷基化历程需要底物由能量较低的1-氢构型转变到较高的2-氢构型,需要克服4.46 kcal/mol的能垒,然后才能进行烷基化,将这两步看成一步,N1烷基化所需克服的总的反应能垒为17.22 kcal/mol,比N2烷基化高出3.35 kcal/mol。利用阿伦尼乌斯公式计算可得22 °C下N2和N1产物的比例大约为286比1(图7)[3],与实际反应的结果是一致的。多数情况下,2-氢互变异构体N1烷基化活化能低于1-氢互变异构体N2烷基化。但2-氢互变异构体需要从能量更低的1-氢构型转变过来,从而增加了能量壁垒。N1烷基化产物的能量通常低于N2烷基化产物,这可能是由于空间效应导致的。
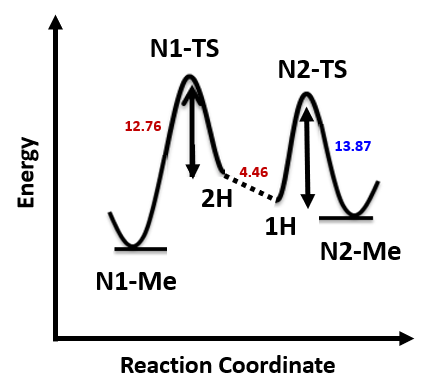
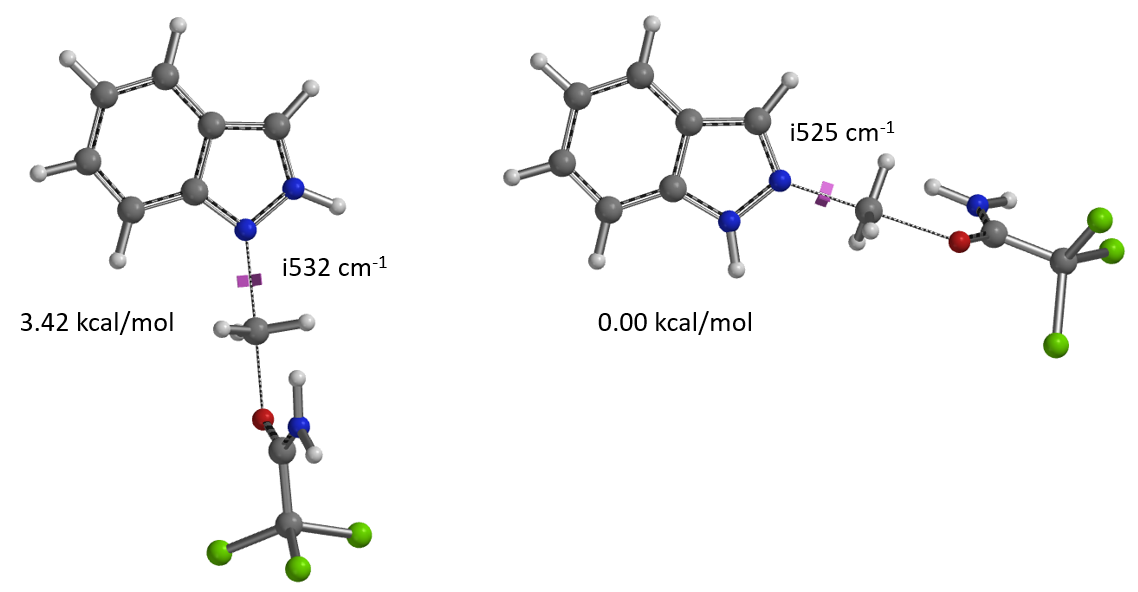
使用这两个过程中能量最高点计算过渡态,分别有且只有一个虚频,验证了这两个过渡态结构的合理性[4]。对比过渡态能量可以看到N1烷基化过渡态比N2高出3.42 kcal/mol的能量(图6),也就是该反应更倾向于发生在N2位。
进一步计算可得25 °C下N2和N1产物的比例大约为322比1,这一分析方法得出的结论与实验结果也是一致的。能量差在3 kcal/mol以上,很难通过调节反应条件逆转反应本身的偏好[3]。即使将反应温度升至101 °C (二氧六环溶剂的沸点),计算N2和N1产物的比例仍高达100比1(图7)。
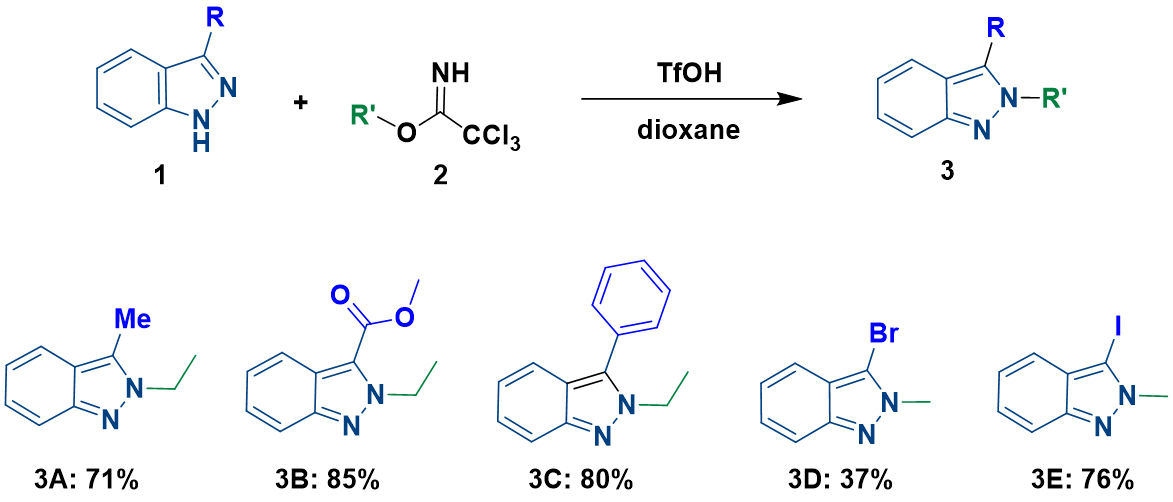
一个有趣的现象是文章给出了几种3-位取代的吲唑底物,尽管3位取代基具有较大的空间位阻作用,但反应都选择性的获得了N2-烷基化产物。我们进一步将该应用拓展底物范围,用3-溴和3-碘吲唑作为底物,在文章标准条件下分别进行了烷基化反应,反应同样均只监测和分离到N2取代的产物。
通过一系列计算,我们得到3位为各种不同取代基时,对应1-氢和2氢互变异构体的相对能量,2-氢吲唑N1、1-氢吲唑N2烷基化的活化能及过渡态相对能量差值。在所有计算的结构中,2-氢互变异构体均为高能构型,较对应的1-氢互变异构体高出4 kcal/mol以上的能量。其中,3-溴吲唑两种互变异构体的相对能量差高达6.99 kcal/mol。图8第6列为综合考虑互变异构体能量差和能量变化曲线得到的N1、N2烷基化活化能之差,多数情况下N1烷基化所需活化能更高,但有三个底物情况相反。互变异构体之间相对能量的估算不太精确,可能会影响分析结果的准确性[5]。相比之下,计算过渡态能量是一种更简单、更可靠的方法。对比过渡态能量(图8第7列),我们可以看到所有底物N1烷基化都是相对较高的,除3-叔丁基吲唑外,要比N2高3 kcal/mol以上的能量,可以很好的解释为什么反应更倾向于发生在N2位。
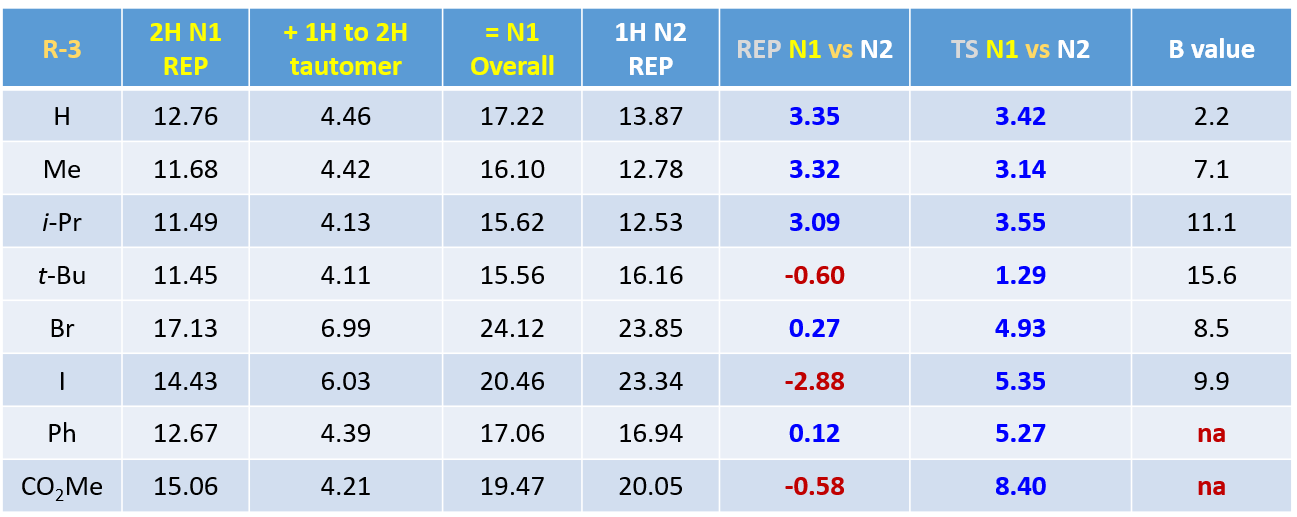
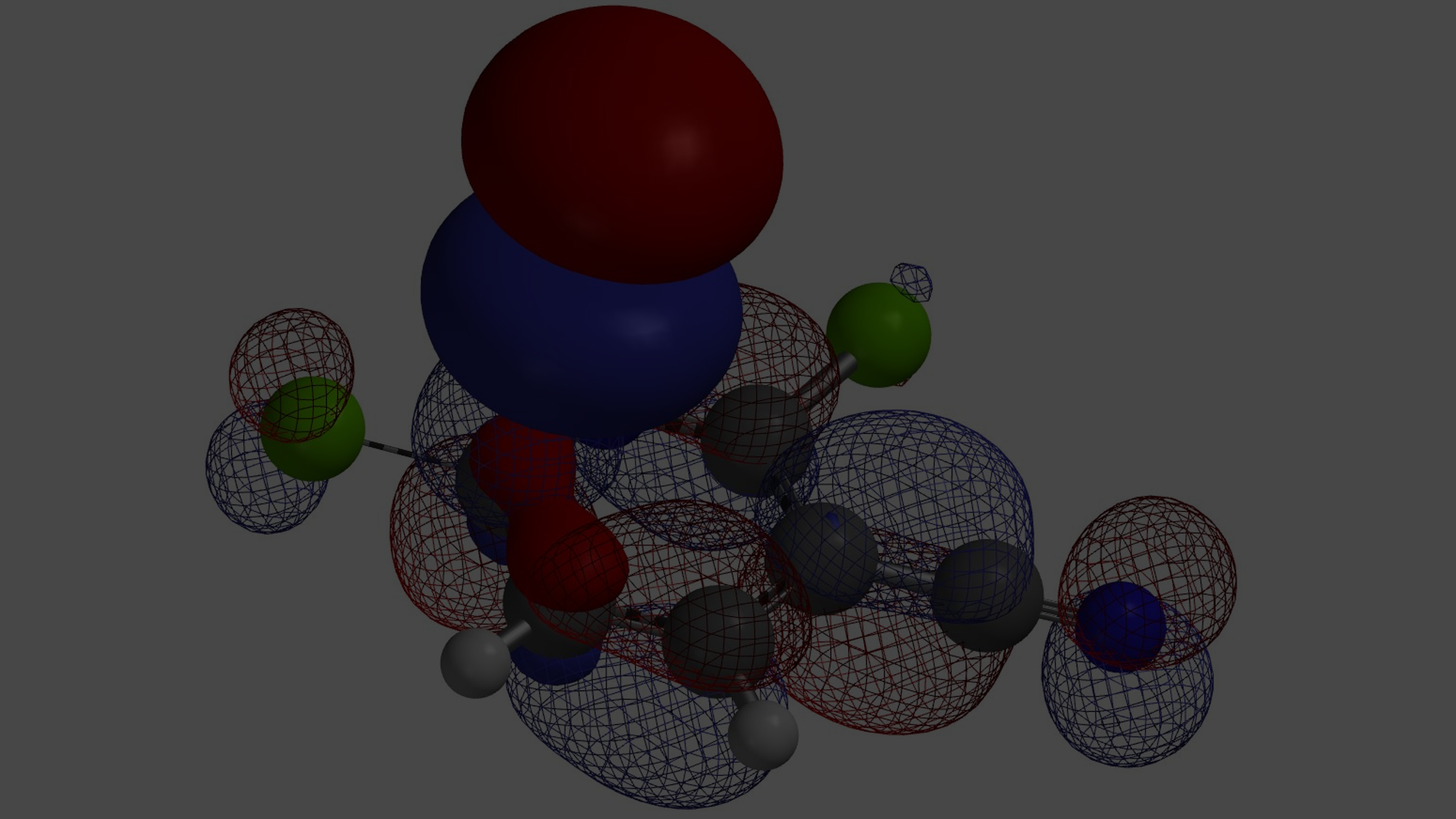
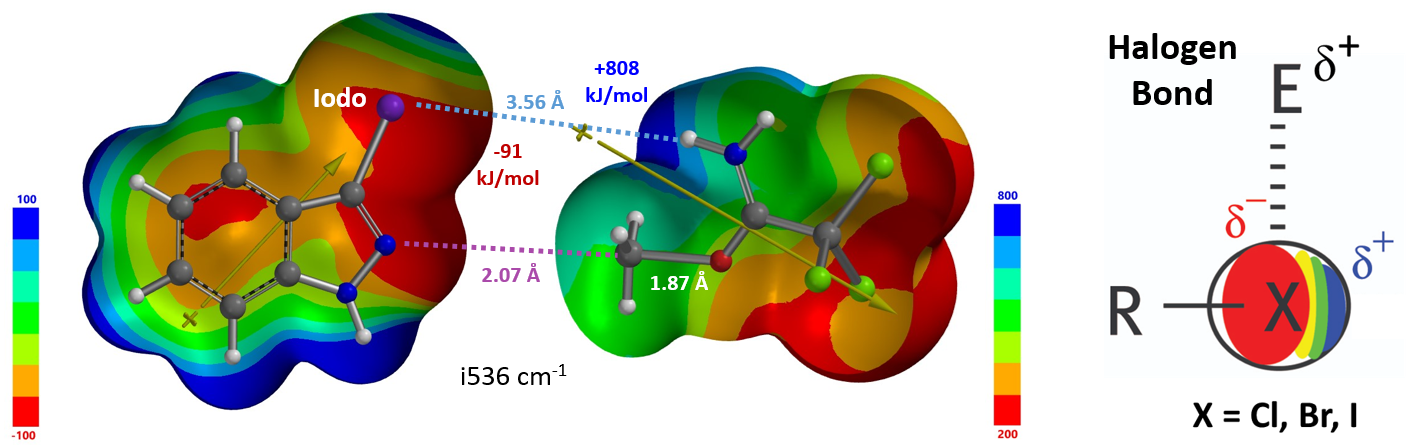
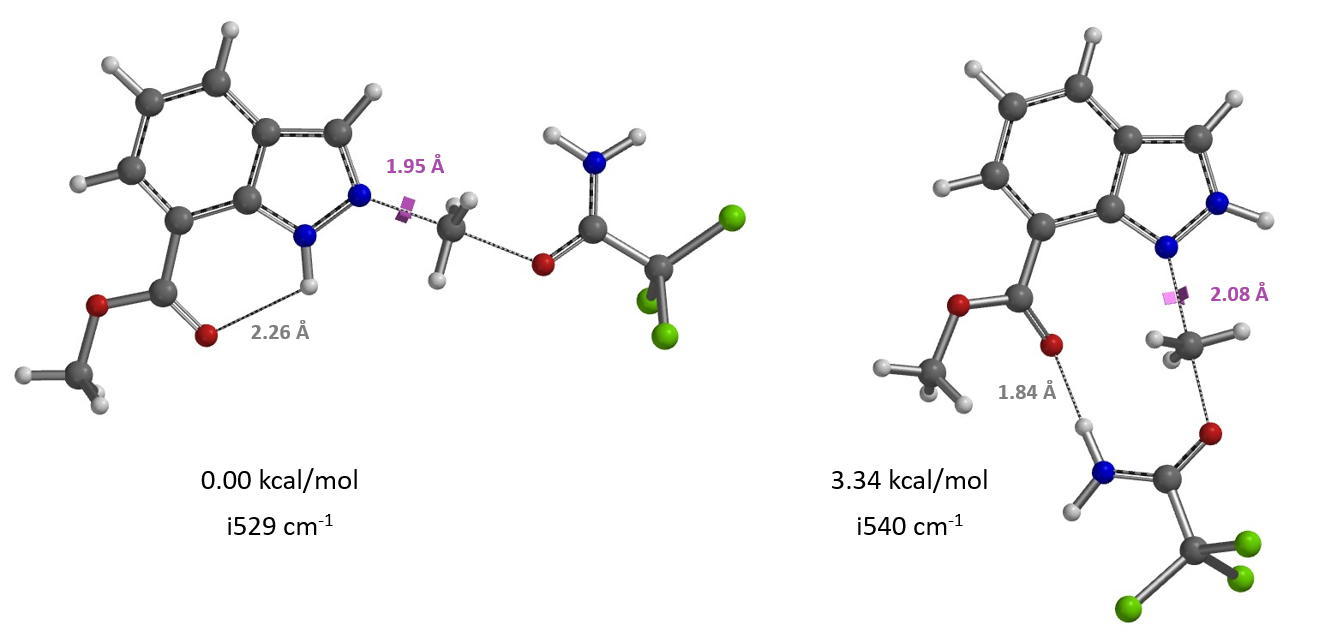
对于简单的烷基,我们可以看到过渡态相对能量与其空间参数有较为合理的相关性(图8中列出的B值)[6]。取代基空间位阻效应越大,对应的B值越大,越不利于N2反应进行,N1与N2烷基化过渡态能量差相应减小。对于3-溴和3-碘代物,如果它们表现得像简单的烷基,基于B值其对应N1与N2过渡态能量的差值应约小于 4 kcal/mol,而不是大约5 kcal/mol。一个合理的解释是3-溴或碘吲唑与质子化亚胺酯之间存在某种较强的相互作用,稳定了N2烷基化的过渡态,从而导致N1、N2过渡态能量差变大。如图9左所示,计算发现3-碘吲唑的碘原子与质子化亚胺酯的亚胺氢质子间存在非常强的静电相互作用,类似于卤素键(图9右)[7],从而大大降低了N2过渡态的能量,最终导致了过渡态能量差与空间参数B值不一致的现象。
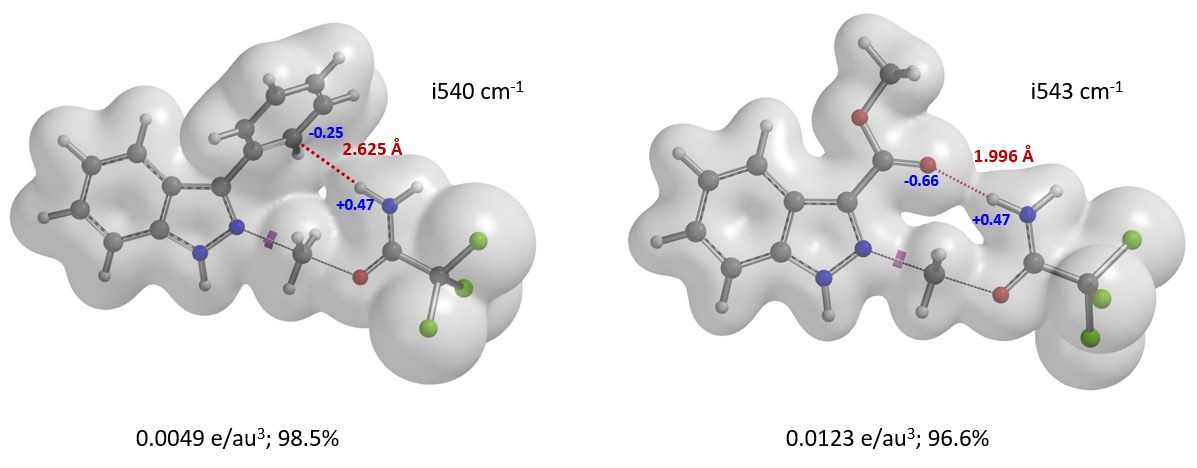
文献没有报道苯基和甲氧羰基的B值,计算揭示了苯基和酯基底物N2烷基化过渡态中均存在非共价相互作用。如图10(左)所示,3-位苯基的一个碳原子与亚胺氢质子间的电子密度表面像一座桥一样连接起来[9],表明两原子间可能存在库伦相互作用,从而降低了N2烷基化过渡态的能量。吲唑-3-羧酸甲酯,N1、N2过渡态能量差值高达8.4 kcal/mol。究其原因是因为N2过渡态中酯基的羰基氧原子与亚胺氢质子间存在较强的氢键相互作用(图10,右)。这种氢键作用降低了N2烷基化过渡态能量,加强了反应对N2烷基化的固有“偏爱”。
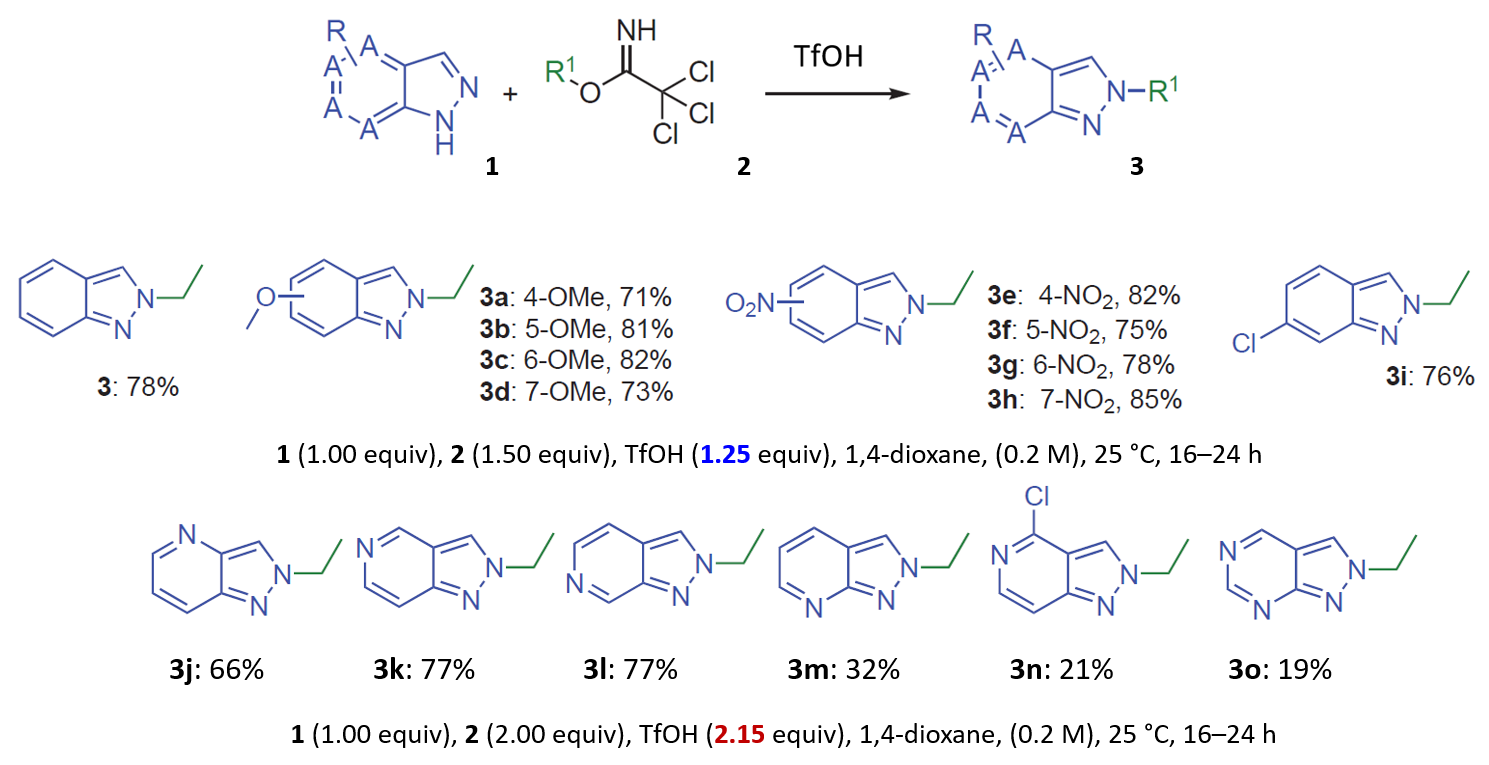
对于氮杂吲唑类底物,文章提到使用1.25当量的TfOH观察到有限的或没有反应发生,当TfOH增加到2.15当量、升温至60 °C时则会选择性的生成N2烷基化产物。作者推测,添加的酸优先使更碱性的吡啶/嘧啶氮质子化,从而增强N2氮的路易斯碱性[1],一个相当奇怪的解释。我们决定使用以上计算模型来解释这些结果。
首先,我们来试着分析一下为什么使用1.25当量的TfOH氮杂吲唑烷基化难以发生。这时反应有两种可能的历程,TfOH质子化活化亚胺酯后与氮杂吲唑反应或质子化氮杂吲唑后与未活化的亚胺酯反应。我们假设如吲唑类底物一样,被TfOH质子化的是亚胺酯。前面我们使用Spartan (DFT wB97X-D/6-31G*) 计算了吲唑N2甲基化反应的能量变化曲线,得出其所需活化能为13.87 kcal/mol。用同样的方法可以计算得到氮杂吲唑N2甲基化所需活化能为14.89 kcal/mol,两种反应的活化能很接近,差值只有大约1 kcal/mol,这显然与实验现象不一致(图11)。
我们通过计算发现质子化吡啶氮对应的能量更低,二者能量相差高达9.54 kcal/mol。因此,可以断定TfOH 第一个质子化的是在碱性更强的氮杂吲唑的sp2氮原子。
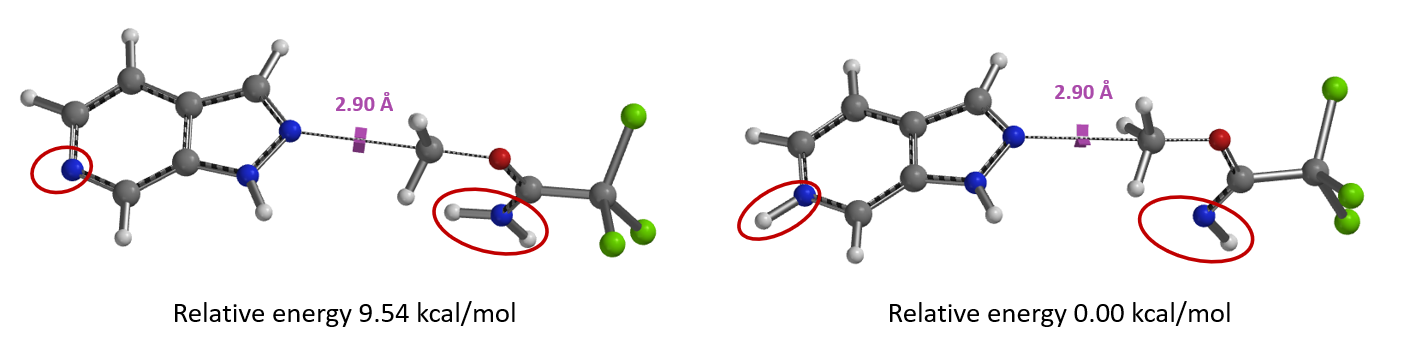
计算质子化氮杂吲唑和未活化亚胺酯烷基化反应的能量变化曲线(图13,右),所需活化能高达39.98 kcal/mol,远大于吲唑的13.87 kcal/mol,很好的解释了为什么两种底物反应结果差异如此之大。吡啶氮被质子化后吲唑反应活性降低而亚胺酯未被活化,反应能垒较高,所以使用1.25当量TfOH时反应难以进行。
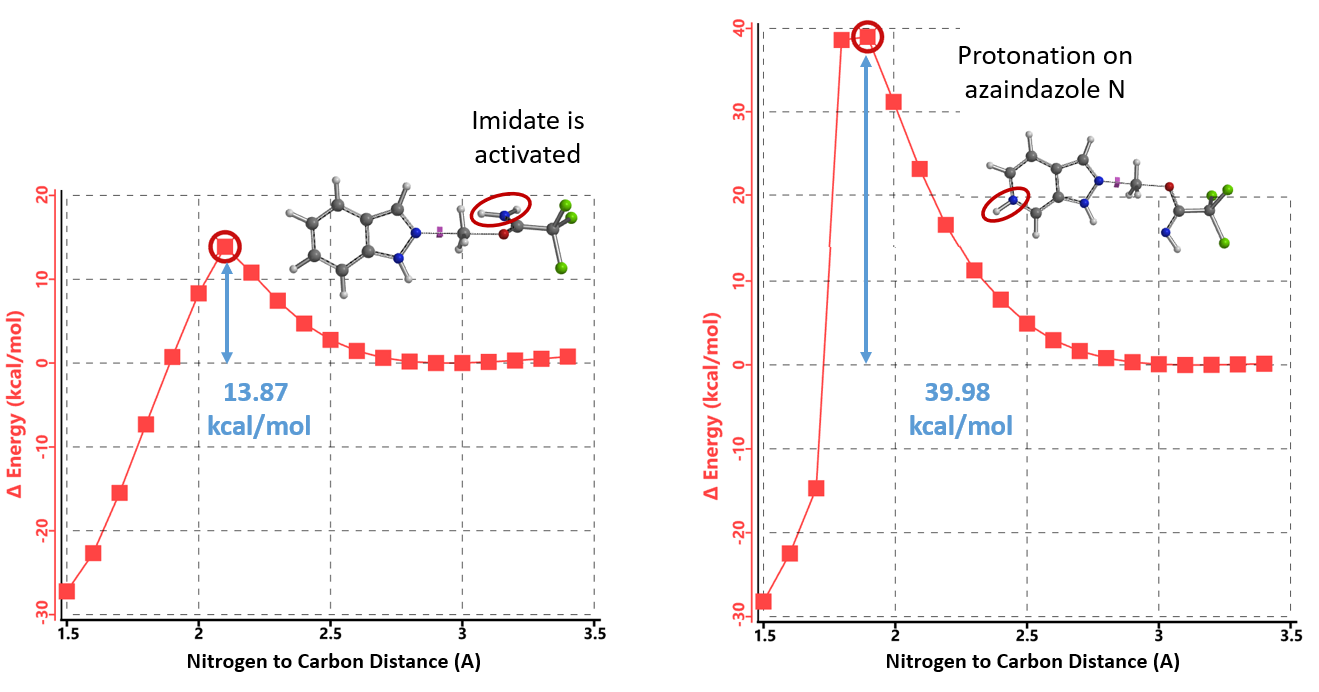
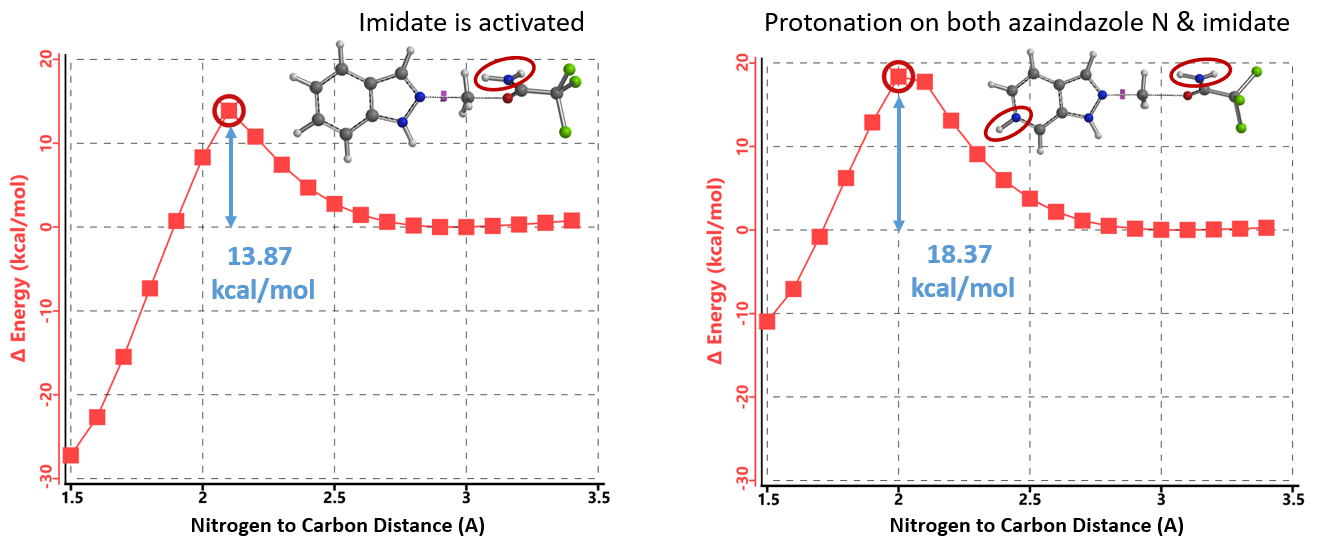
当TfOH的用量增加到2.15当量时,氮杂吲唑和亚胺酯都能被质子化,计算相应的能量变化曲线(图14,右),反应所需的活化能为18.37 kcal/mol,属于实验条件下可克服的合理的能垒。氮杂吲唑质子化后活性降低,而被活化的亚胺酯仍具有足够的反应性,可以使质子化的氮杂吲唑甲基化,但所需活化能相对较高(较吲哚高4.5 kcal/mol),反应需要增加亚胺酯的当量(从1.5增至2.0当量)和升温来提高转化率。由此可见,文章中作者提出的“添加的酸优先使更碱性的吡啶/嘧啶氮质子化,从而增强N2氮的路易斯碱性”的假设明显是不成立的。
本章我们利用QM工具计算验证了吲唑高选择性N2烷基化的反应机理,通过比较过渡态相对能量说明了为什么吲唑环3位取代基不会显著影响反应的选择性,并推导验证了氮杂吲唑与吲唑烷基化反应条件差异化的原因。
关于吲唑的环状互变异构化,已经进行了广泛的计算和经验研究。1-氢吲唑互变异构体在能量上占主导地位,多数情况下ΔE298 (1H→2H) 大于4.0 kcal/mol,对于互变异构体参与的转化要考虑到其相对能量差异。值得注意的一点是,互变异构体相对能量的估算不太精确,可能会影响分析结果的准确性。相比之下,计算过渡态能量是一种更简单、更可靠的方法,可更有效的用于与实验观测结果进行比较和关联。
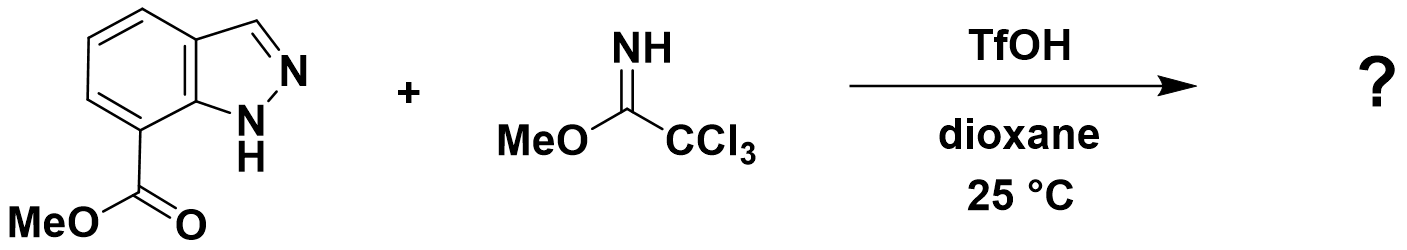
接下来,请大家思考一个问题,你能根据图15及16提供的信息预测吲唑-7-羧酸甲酯烷基化反应会发生在哪个位点吗?欢迎大家在评论区留言。
本文由董立亭、王秋月、卫小文编撰。
参考文献:
[1] J. Clemens, E. L. Bell, A. T. Londregan, Synthesis 2022, 54, 3215.
[2] 辉瑞的化学家们认为,当R是一级或二级烷基时,反应通过SN2反应机制进行; 当R为三级烷基时,考虑到三级烷基的立体位阻效应反应可能通过三级烷基阳离子进行。
[3] 量子力学有机化学第九章《利用活化能计算预测吡唑氮烷基化的区域选择性》。
[4] a) Spartan'20 Tutorial and User's Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp. 158, 442, 459 & 536. b) 量子力学有机化学第二十二及二十四章有关过渡态和虚频的计算。
[5] 量子力学有机化学第四十九章《互变异构体》。
[6] R. Ruzziconi, S. Spizzichino, L. Lunazzi, A. Mazzanti, M. Schlosser, Eur. J. Chem. 2009, 15, 2645
"B values as a sensitive measure of steric effects.”
[7] P. J. Costa, Phys. Sci. Rev. 2017, 2, 136.
[8] 为了更清楚的显示静电势图,吲唑和亚胺酯被人为的拉开了一定的距离,且两个片段的静电势图以不同的比例静电势显示。
[9] 量子力学有机化学第四十二章《利用电子密度图实现氢键可视化》。
