












网站维护
系统内容更新/升级中


各位《QM魔法小课堂》的粉丝们,我们又开讲了。
在上一期的理论课里,我们提到了利用哈蒙德假说,可以构建反应过渡态的结构,计算反应活化能,辅助预测化学反应。小伙伴们是不是都很好奇,想在实际工作中演练一下呢?
我们来看下面这个例子:化合物1的烷基化反应(图一),该烷基化反应会得到的是1位还是2位的烷基化产物呢?如果得到的是混合物,两者的比例又会怎么样呢?
带着这两个问题,我们首先分析一下该反应的进程:碱性条件下,吡唑NH去质子转化为N负离子,之后发生烷基化。
利用第三章学习的静电势图,可以比较吡唑的两个异构体中,NH的酸性强度,判断哪个NH会优先和碱反应形成负离子(图二A和B)。异构体A中的NH静电势(281 kJ/mol)略高于异构体B中的NH静电势(266 kJ/mol),但差异并不显著;而且在这个案例中这点也并无太大意义,因为NH去氢后,负离子在两个氮原子上是离域的,因此负离子状态下,两个N原子上的HOMO lobe大小也非常接近(图二C)。
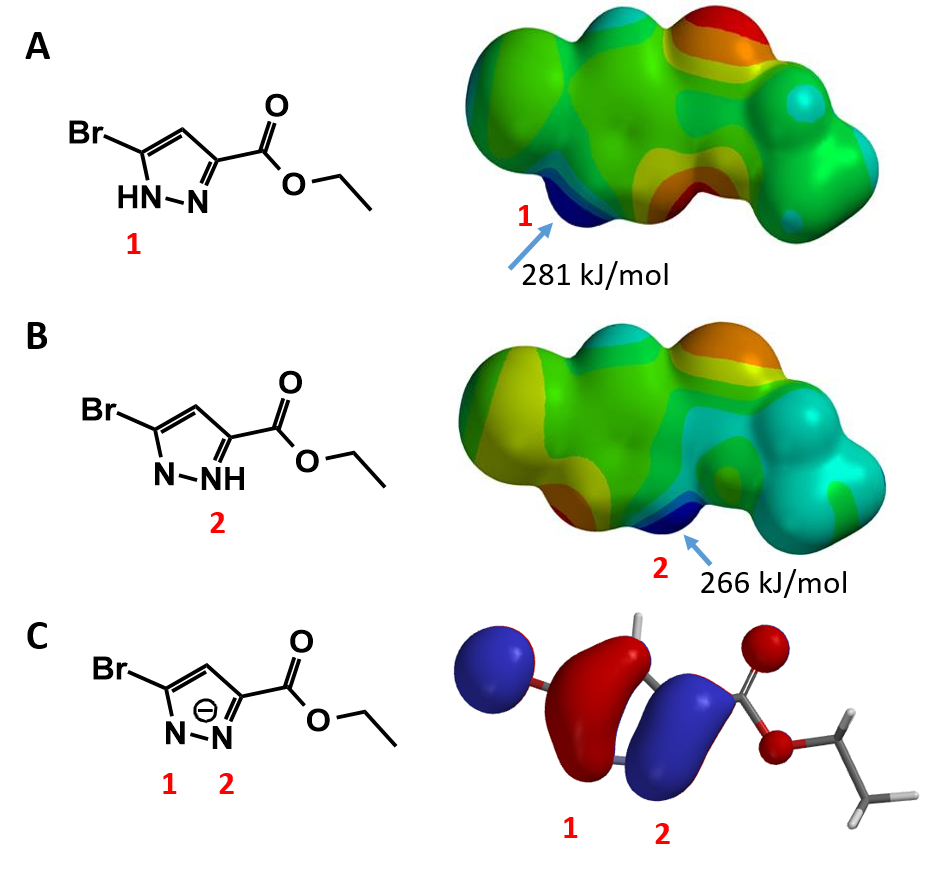
所以,在这个案例中,仅凭静电势图和HOMO分布难以判断反应的选择性。同时,由于两个氮原子的邻位分别有不同的取代基,会带来不一样的电子效应和位阻效应。因此我们需要进一步分析,来预测产物情况。
现在,有请本章节的主人公——活化能计算(Reaction Energy Profile),隆重登场!
活化能计算的基本概念是通过QM模拟反应成键或断键的过程,寻找反应过渡态,得到反应的活化能值,利用活化能的大小判断反应的难易,并利用活化能差值计算出不同产物的比例。
再来看我们的例子,吡唑化合物1的烷基化反应为典型的SN2反应,其机理为N负离子逐渐接近卤代烷烃,形成C-N键,同时发生C-X键的断裂。
以CH3Br为反应物,我们分别在CH3Br的C原子与化合物1的N1或N2原子之间模拟C-N键的形成,即定义C-N之间的距离,从彼此无相互作用的远距离3.8 Å,逐渐接近至进入共价键作用范围的1.9 Å,计算这一过程中,系统能量的变化(图三)。
我们发现C-N原子间距离缩短到3.0 Å时,系统能量开始上升,说明反应物分子开始相互作用。键长缩短至2.1 Å左右时,能量达到最高点,反应物分子形成了过渡态络合物。之后C-N之间逐渐形成稳定的共价键,C-Br键断裂,系统能量降低,烷基化反应趋于完毕。
通过计算,我们发现N1位置发生烷基化反应所需的活化能约为10.77 kcal/mol,N2位置发生反应的活化能约为9.24 kcal/mol(图三)。N1位烷基化所需要的活化能比N2位的要高,说明反应更容易发生在N2位上。
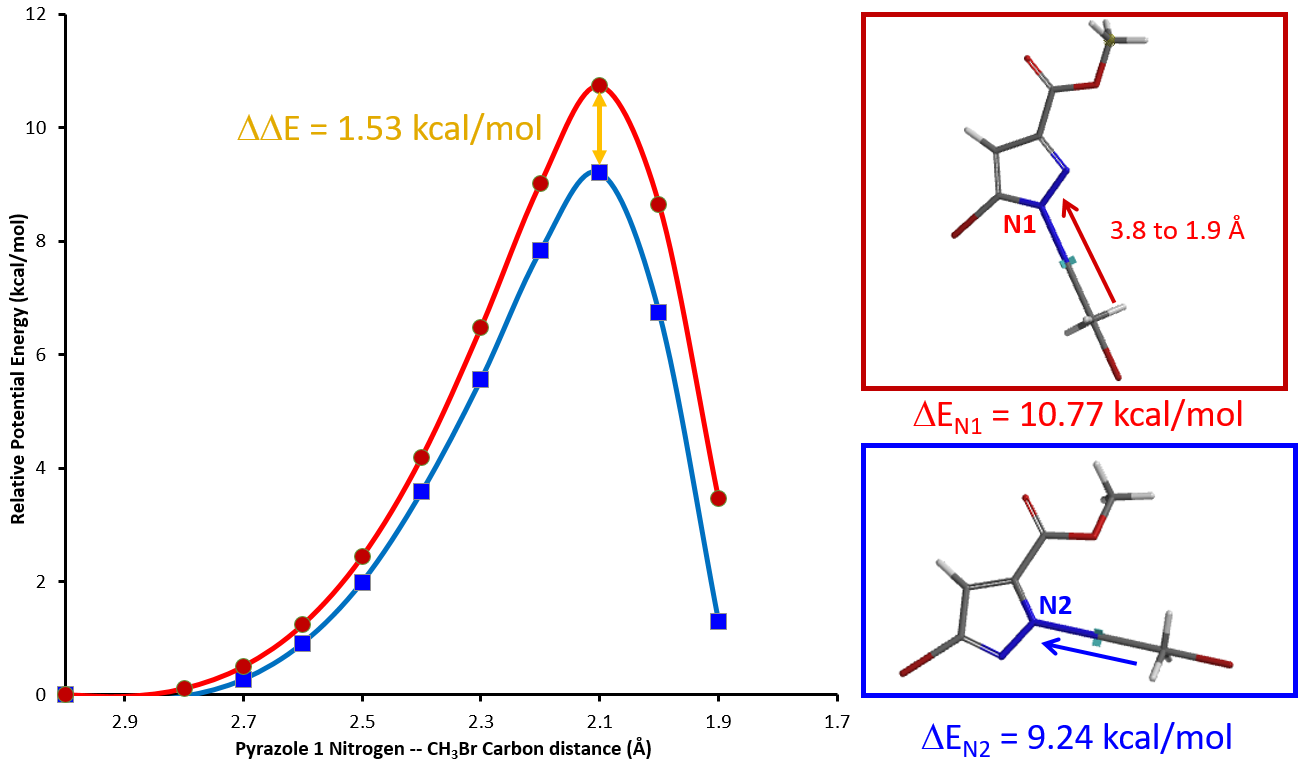
根据两个反应活化能的差值,我们可以进一步利用阿伦尼乌斯公式(图四)定量计算两个产物的比例(具体推导可详见第八章《站在巨人的肩膀上,哈蒙德假说》)。根据图三,N1与N2位烷基化反应的活化能差值 ![]() 为1.53 kcal/mol,代入图四公式后,可以计算得到25℃时,N2位产物与N1位产物的比例约为13:1。
为1.53 kcal/mol,代入图四公式后,可以计算得到25℃时,N2位产物与N1位产物的比例约为13:1。
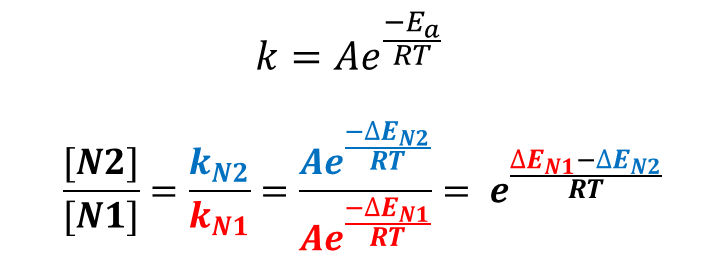
有些小伙伴们可能觉得数学已经离自己很遥远了,看到这样的公式有点眩晕。
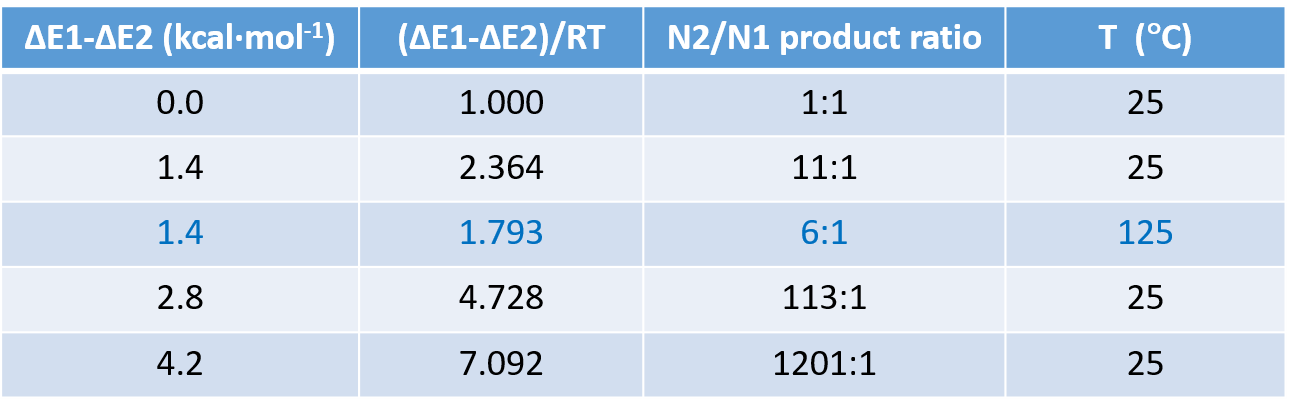
这里我们总结出一个表格(表一),只需代入两个反应的活化能差E,简单对比一下表格的数据,就可以得到两个产物的大概比例了,是不是方便了很多?
当然,影响反应速率的因素分为内因与外因。
内因主要是反应物的性质,以及不同的反应位点和环境,需要不同的活化能。除此之外,外因也是影响反应的重要因素,如温度、溶剂、浓度、压强、催化剂等。
如表一所示,当反应的活化能差值为1.4 kcal/mol时,如果反应温度为25℃,类似室温条件时,两个产物的比例为11 : 1;当提升反应温度到125℃,类似加热条件时,两者的比例则下降为6 : 1。
我们在实践中发现,当活化能差值在1.4 kcal/mol附近时,我们可以根据经验,调节温度、溶剂等外部条件,控制产物的比例,以获取我们的目标产物。如果计算预测的活化能差值在3 kcal/mol以上甚至更高,那原产物比例则高达100 : 1及以上,逆转反应本身的偏好将非常困难,此时就需要调整思路,利用不同的反应机理或者重新规划反应顺序,以获得目标产物。
理论是要通过实践来检验的,那么具体的实验的结果是什么呢?我们在25℃下反应时,发现N2烷基化和N1烷基化两种产物的比例约为10 : 1,和我们计算的结果很接近!
总结一下,通过模拟C-N键的成键过程,我们找到了反应的过渡态,进而得到了反应活化能的差值,计算得到产物的比例,成功预测了吡唑氮烷基化的选择性偏好。
怎么样?通过本章节的学习是不是对活化能计算的应用有了一定的了解?结合QM的计算,不仅能帮助我们深入地理解和印证课本中学习的理论,还能将这些理论与计算,变成我们合成人员手中一个强有力的工具。大家记得一直来学习探讨哦!
本期结束,大家有没有跃跃欲试,看看这么高大上的理论能不能在自己手里玩转呢?这里也有一个例子供大家课后思考与练习:
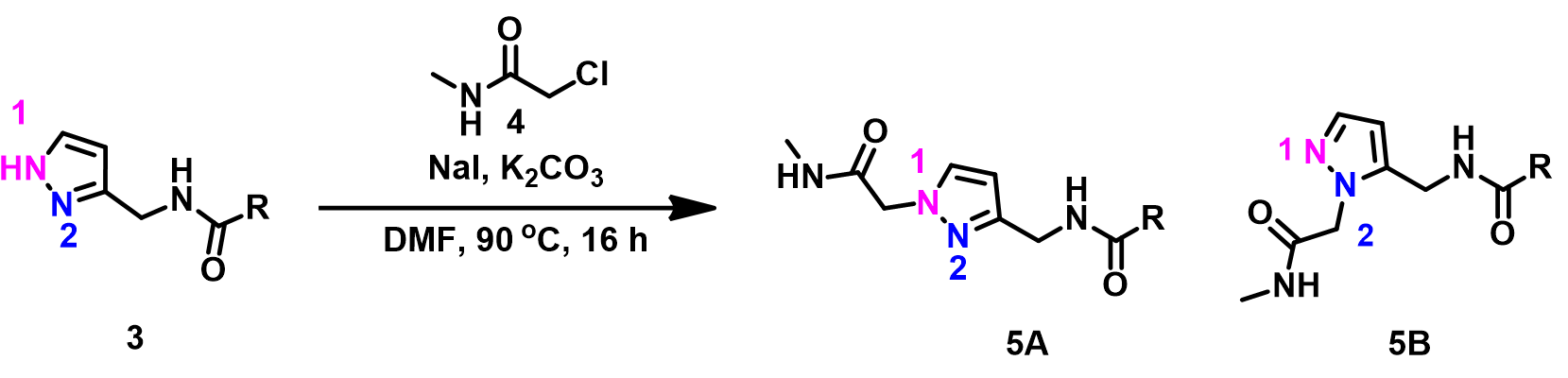
同样是吡唑类化合物3和卤代乙酰胺化合物4的烷基化反应,反应发生的位点会在哪里呢?两个底物中均出现了活泼氢,这些活泼氢会对反应的选择性产生影响吗?
温馨小提示:
接下来的章节,我们将进一步介绍如何运用本章所学的活化能的知识,来解决更多的化学问题。敬请期待
本文由王守亮、王坚、石谷沁、王秋月、潘东、卫小文编撰。
参考文献:
[1] Warren J. Hehre (2003). A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc.
[2] Spartan '18 Tutorial and User's Guide (2019). Irvine, CA, USA: Wavefunction, Inc. p116 - 118
[3] S. Liu, L.G. Pedersen. J. Phys. Chem. A. 2009, 113, 3648
[4] 杨宏孝, 王建辉. 无机化学. 第四版. 北京: 高等教育出版社., p270-279
