












网站维护
系统内容更新/升级中


多卤素化合物在不同类型反应中的区域选择性一直是困扰大家多年的谜题。如图1所示,2,5-二溴吡啶的芳香亲核取代反应发生在2位[1](亲核取代选择性预测见第1章、第7章、第10章、第19章),钯催化的Buchwald偶联反应也是2位的溴优先反应[2](偶联反应选择性预测见第5章),但是卤素金属交换反应的区域选择性却完全不同,5位溴原子优先反应[3, 4]。
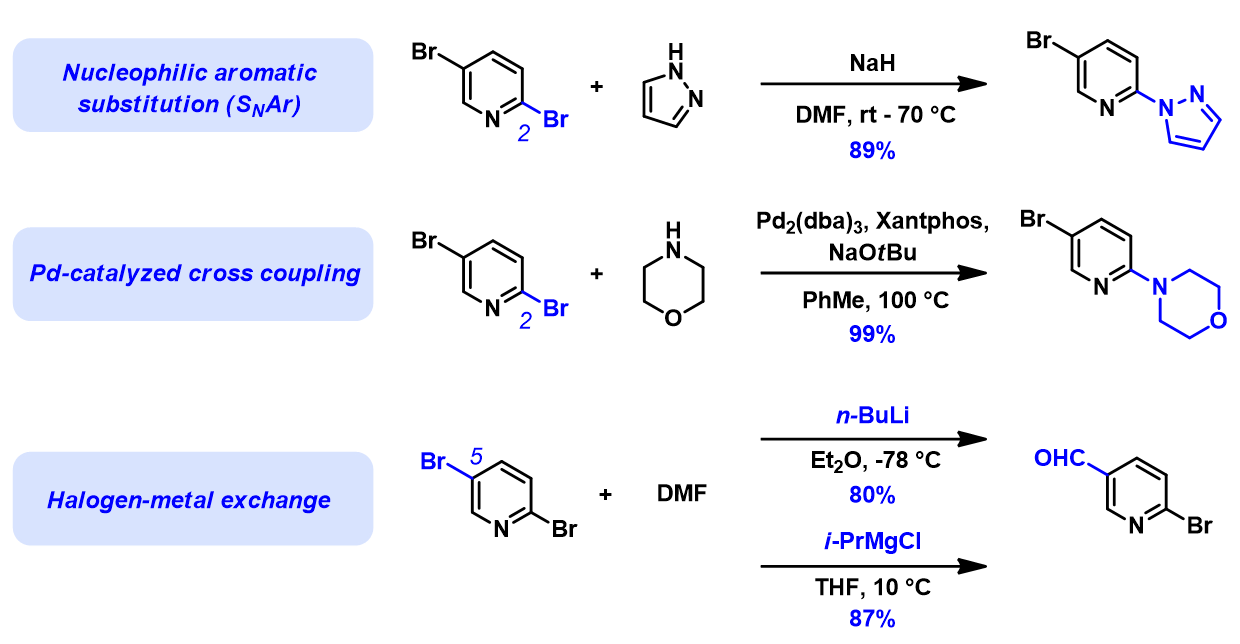
除此之外,还有许多其他的案例,多卤素化合物在亲核取代、偶联和卤素金属交换反应中表现出区域选择性的差异[5, 6]。数十年来,大家一直在寻找这个问题的答案:这种选择性差异的原因是什么呢?
QM作为一项强大的工具,能够帮助我们拨开重重迷雾,看清这些差异背后的真相吗?
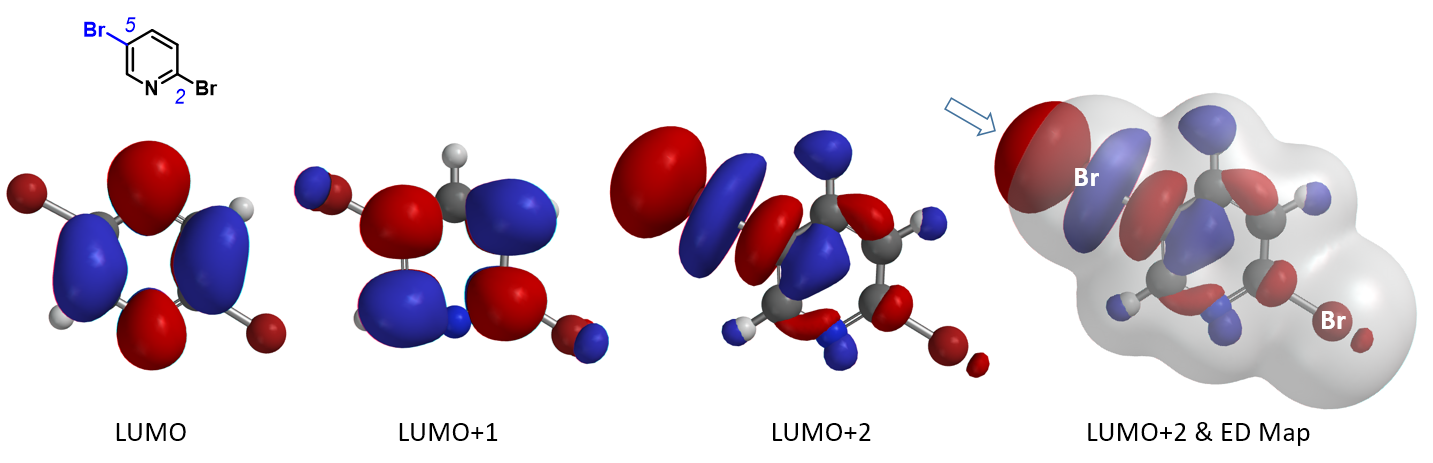
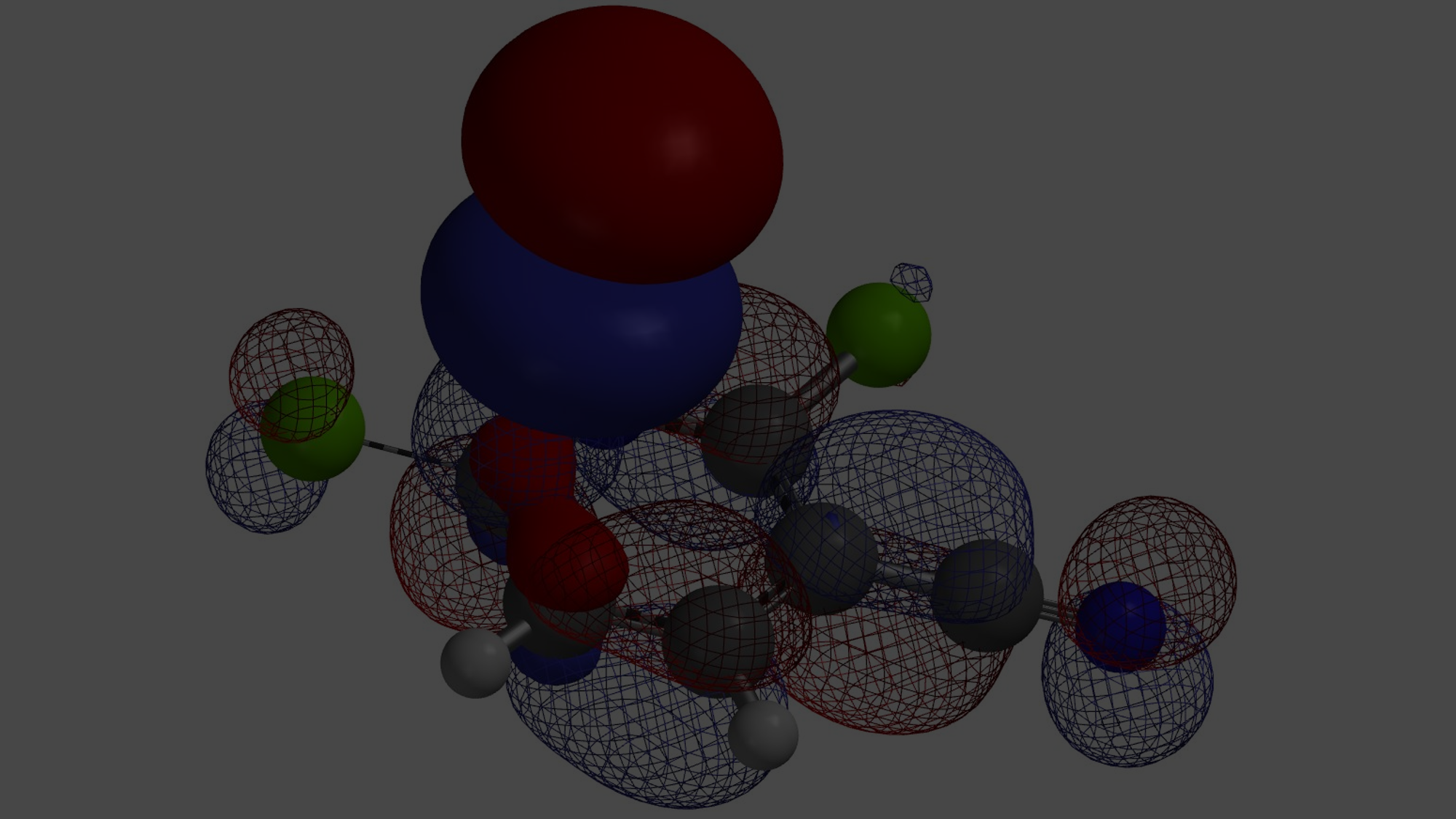
接下来,我们将这个方法应用到更多的案例中(图4)。1,2,4-三溴苯的2-位溴上有体积最大的LUMO+2 lobe分布,与Turbo试剂反应选择性得到2-位取代产物。2,3-二溴噻吩的2-位溴与格式试剂优先反应,2-位溴的LUMO+1 lobe分布明显更大。N-甲基-3,5-二溴吡唑的LUMO显示5位溴的lobe分布明显大于3位溴。N-甲基-5,7-二溴吲哚的LUMO+2 lobe显示7位溴上的lobe体积明显大于5位溴,实验结果也都是lobe分布体积更大的溴原子优先发生卤素金属交换反应。
最后两个案例代表了卤素-金属交换反应的独有特征,就是邻近的甲基基团对反应的位阻影响很小,与之相对应的则是位阻对偶联和SNAr反应影响明显。这种远远超出Electron Density Map的特殊LUMO+n lobe,就可以很好解释卤素金属交换反应对位阻不敏感的原因。
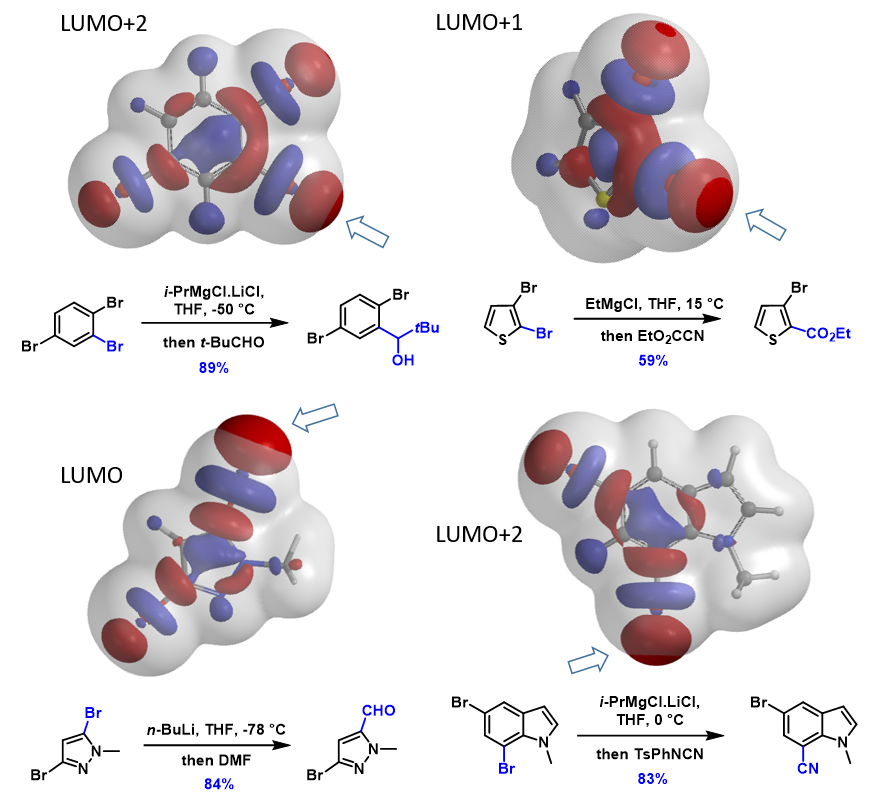
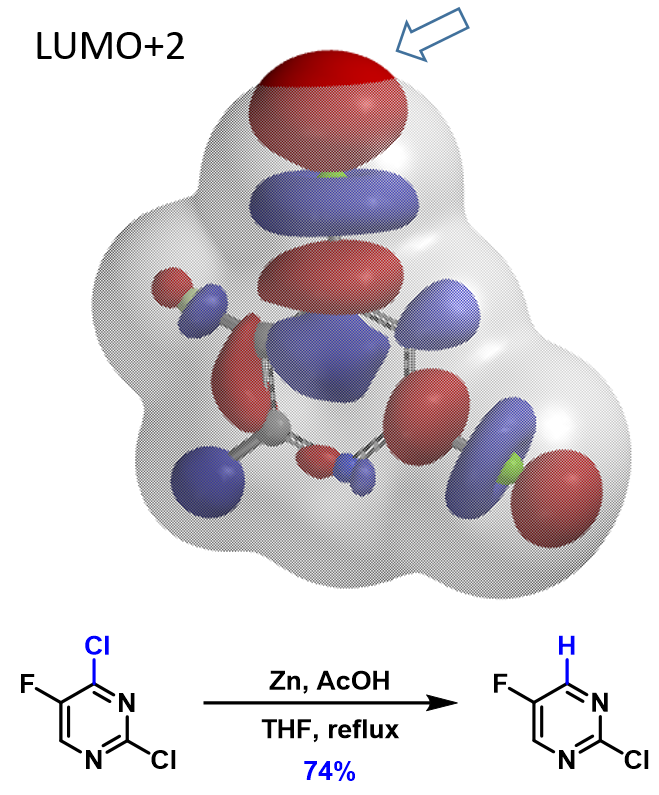
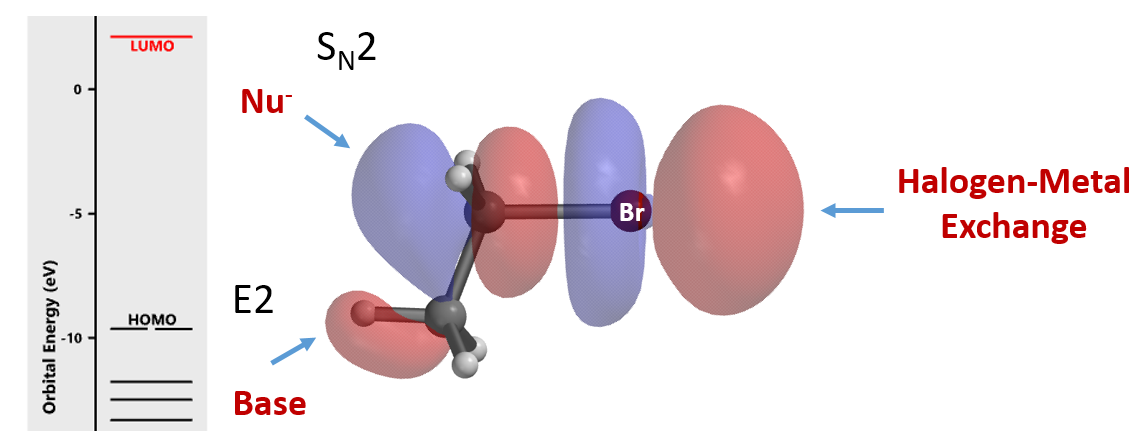
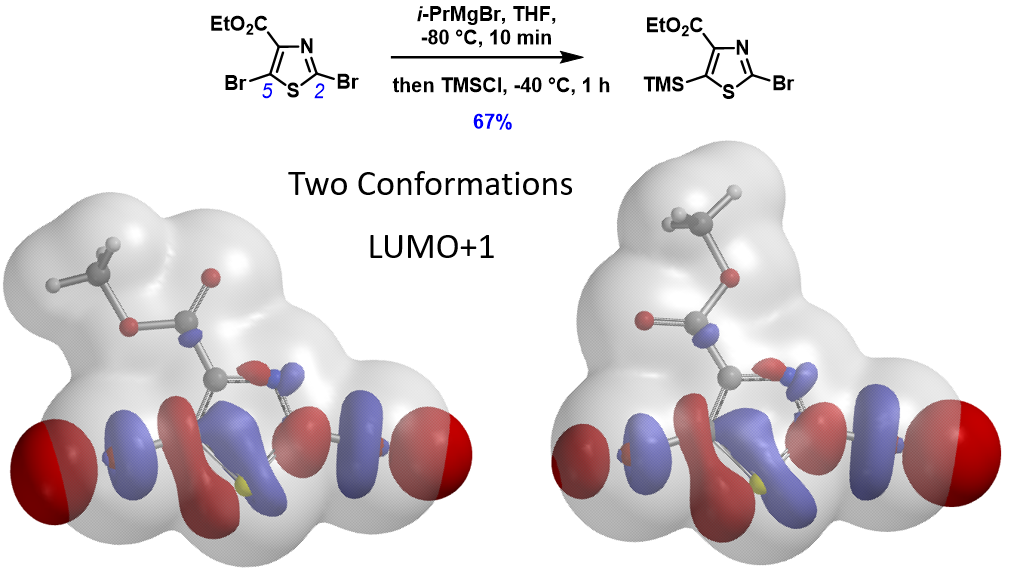
对于卤素-金属交换反应,利用分布在C-X键上独具特色的4颗“珍珠串”形的LUMO lobe 或LUMO+n lobe,可以让我们准确预测卤素的区域选择性。除了以上6个案例之外,我们还使用该LUMO分析的方法验证了文献报道的100+不同案例。
你可能会有疑问LUMO+2和LUMO+3的轨道能量这么高也可以参与反应吗?当然可以,我们需要考虑的是反应物的HOMO-LUMO能量差(第25章)[8]。n-BuLi和格式试剂的反应活性非常高,对应的HOMO能量也非常高,所以参与反应的LUMO+n轨道能量和HOMO的能量差还在可以发生反应的范围之内。n-BuLi具有比格氏试剂更高的HOMO能量,这也与丁基锂可以在更低温条件下发生卤锂交换的观察一致。
当计算得到的LUMO lobe或 LUMO+n lobe大小接近时,可以进一步比较不同位点反应后形成的芳基金属试剂的相对能量,能量越低,越稳定,在产物中的比例就越高[9]。
虽然真实环境体系中的金属试剂中间体是以碳金属键的形式存在,同时稳定性受溶剂、温度、导向基等各种因素的影响,但是通过简化结构为相应的碳负离子,我们发现计算碳负离子的相对能量也可以很好的预测反应的选择性。
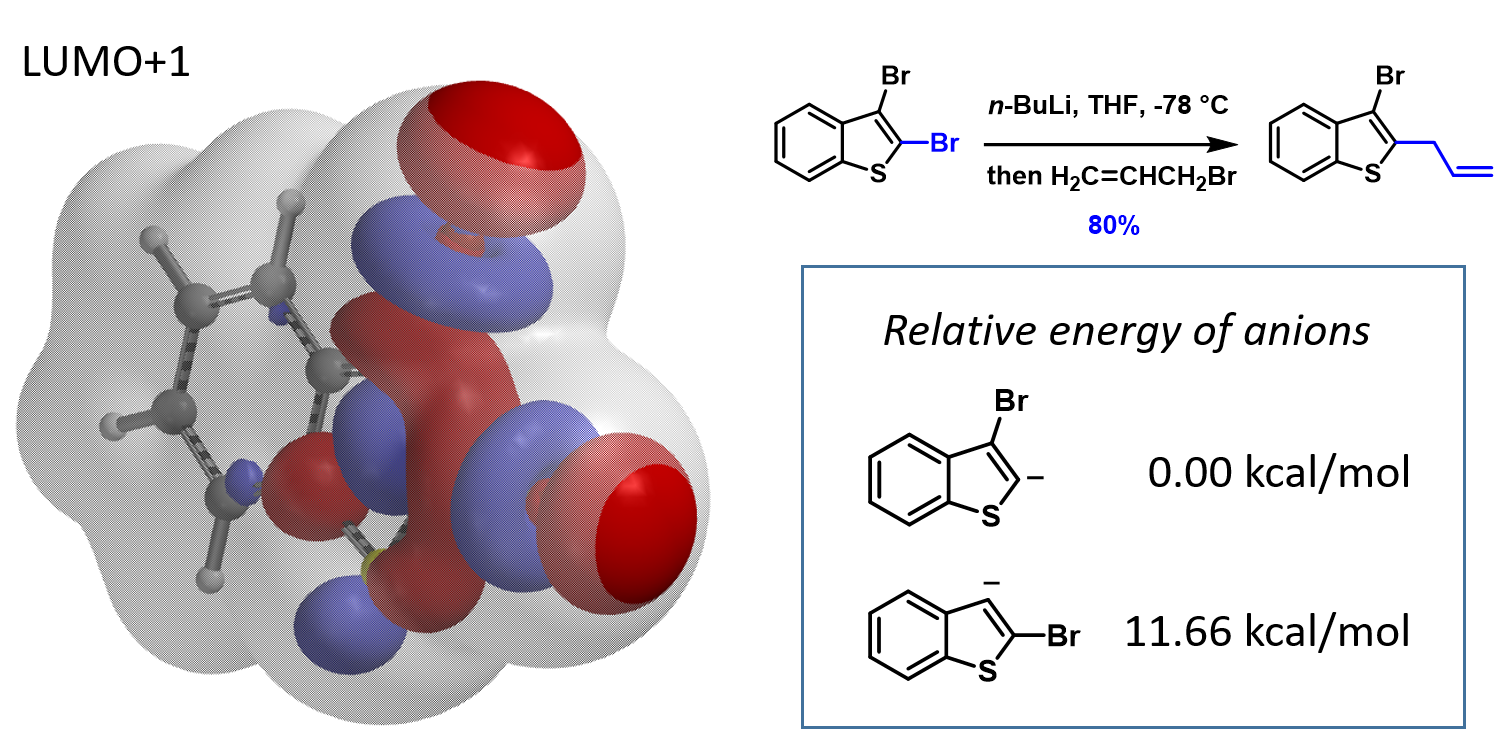
总结一下,金属试剂与C-I、C-Br或C-Cl键上独具特色的“珍珠串”形的 LUMO lobe相互作用发生卤素金属交换反应,通过比较卤素原子上这种独特lobe体积的大小就可以让我们准确预测卤素金属反应的区域选择性。这种独特的lobe分布可能是在LUMO上,也可能是在LUMO+1、LUMO+2和LUMO+3上。当LUMO lobe 体积大小接近时,可以进一步计算相应的碳负离子相对能量,碳负离子能量越低,就越稳定,产物的比例越高。氧化加成和SNAr与格氏试剂和锂卤交换的卤素选择性差别,可以通过化学相互作用的量子性质完美解释。前者使用分布在C-X键碳上的LUMO lobe反应,后者使用“珍珠串”形LUMO lobe,它具有非常大的lobe突出于卤化物的Electron Density Map表面,决定了观察到的选择性。
通过将计算LUMO和碳负离子相对能量结合,可以帮助我们准确预测多卤化物发生卤素金属反应的选择性,帮助我们解决了困扰多年的谜题!QM工具在帮助我们解决反应问题的基础上,更可以帮助我们将模糊的反应活性可视化、数据化,为未来构建全息的化学世界奠定基础。
本文由潘东、刘军、刘文锋、赖光华、卫小文编撰。
参考文献:
[1] C.S. Rani, A.G. Reddy, E. Susithra, K.K. Mak, M.R. Pichika, S. Reddymasu, M.V.B Rao. Med. Chem. Res., 2021, 30, 74.
[2] J.G. Ji, T. Li, W.H. Bunnelle,Org. Lett., 2003, 5, 4611.
[3] P.N.W.Baxter. Chem. Eur. J., 2003, 9, 5011.
[4] L. Peng, J.D. Wen, C.G. Pan, X.W. Pan, Y. Xie, W. He, Q.F. Lin, X.Y. Zhang (Chongqing Sansheng Industrial Co., Ltd., China), CN 112479991 A, 2021.
[5] J.A. Joule & K. Mills, Heterocyclic Chemistry 4th Ed. Malden, MA, USA: Blackwell Publishing Ltd., 2000; pp 26-45.
[6] https://www.scripps.edu/baran/images/grpmtgpdf/Gutekunst_Apr_10.pdf
Divergences in haloselectivity highlighted with 3,6-dibromoindole (pp 2), 2,3,5-tribromo thiophene (pp 3), 2,5-dibromopyridine (pp 6), in palladium catalyzed cross coupling reactions vs halide metal exchange reactions.
[7] LUMO, LUMO+1, LUMO+2和 Electron Density Map在DFT-ωB97X, 6-31G*下计算. (a) A. Krasovskiy, P. Knochel, Angew. Chem. Int. Ed., 2004,43, 3333. (b) C. Christophersen, M. Begtrup, S. Ebdrup, H. Petersen, P. Vedsø. J. Org. Chem., 2003, 68, 9513. (c) Y. Yin, C.J. Chen, R.N. Yu, L. Shu, T.T. Zhang, D.Y. Zhang, Bioorg. Med. Chem., 2019, 27, 1562. (d) P. Anbarasan, H. Neumann, M. Beller, Chem. Eur. J. 2011, 17, 4217. (e) C. De Savi, A. Pape, J.G. Cumming, A. Ting, P.D. Smith, J.N. Burrows, M. Mills, C. Davies, S. Lamont, D. Milne, C. Cook, P. Moore, Y. Sawyer, S. Gerhardt, Bioorganic Med. Chem. Lett., 2011, 21, 1376.
[8] L.G. Zhuo, W. Liao, Z.X. Yu, Asian J. Org. Chem. 2012, 1, 336.
[9] (a) H.J.S. Winkler, H. Winkler,J. Am. Chem. Soc., 1966, 88, 964. (b) H.J.S. Winkler, H. Winkler, J. Am. Chem. Soc.,1966, 88, 969. (c) H.R. Rogers, J. Houk, J. Am. Chem. Soc., 1982, 104, 522. (d) K.B. Kenneth, S. Sklenak, F.B. William, J. Org. Chem., 2000, 65, 2014.
[10] 碳负离子相对能量在nonpolar solvent状态, DFT-ωB97X, 6-31G*下计算. T.P. Sura, D.W.H. MacDowell, J. Org. Chem., 1993, 58, 4360.
[11] M. Abarbi, J. Thibonnet, L. Bérillon, F. Dehmel, M. Rottländer, P. Knochel, J. Org. Chem., 2000, 65, 4618.
