












网站维护
系统内容更新/升级中


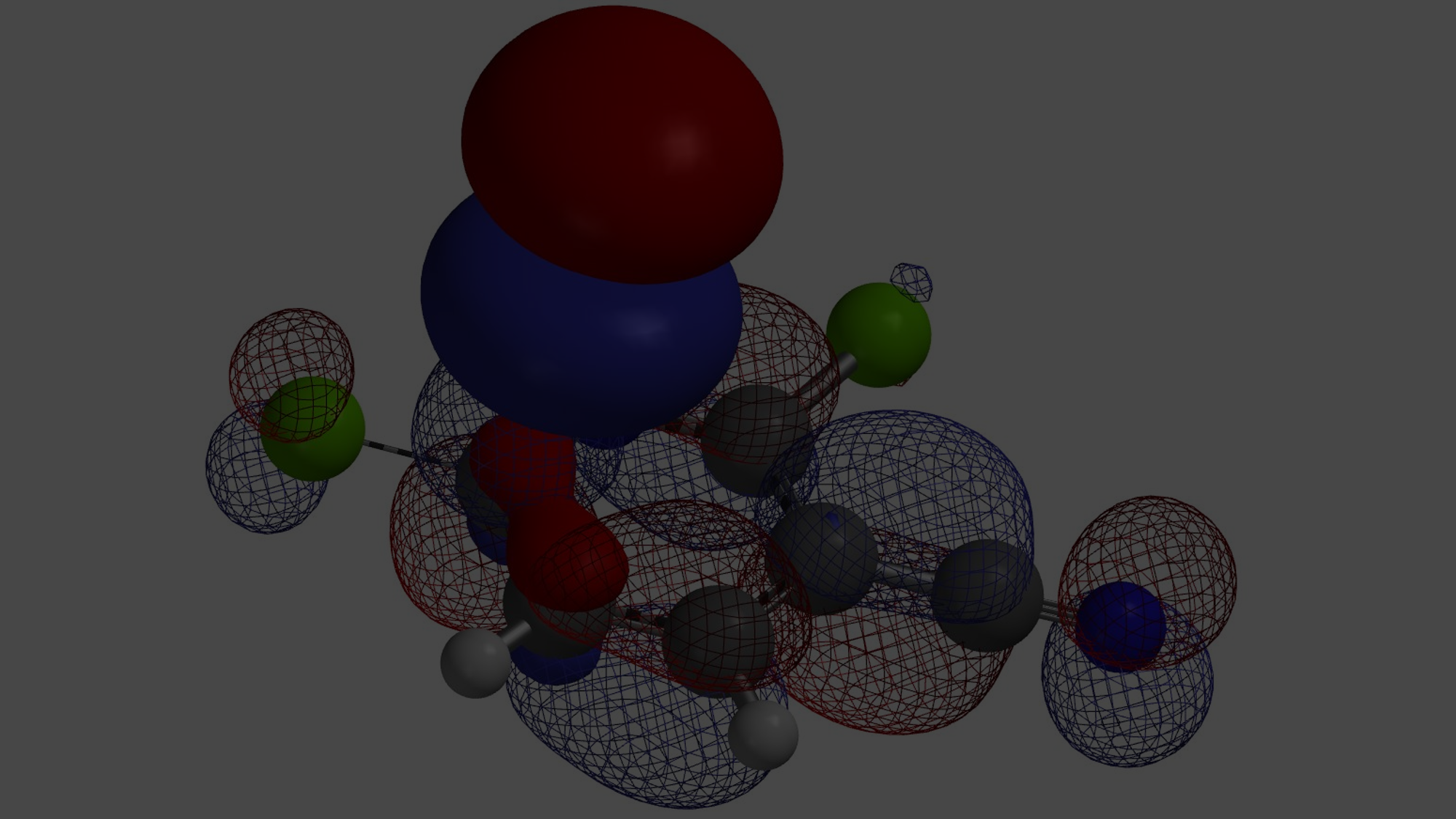
我们在前面的QM小课堂知晓了LUMO及LUMO Map的概念,并通过实例初步了解和学习了它们在有机合成中的应用。本章节我们将介绍运用前面所学内容,为多取代杂环分子设计合适的合成路线。
现在请思考:如何从原料(1)快速合成目标分子(4)呢?
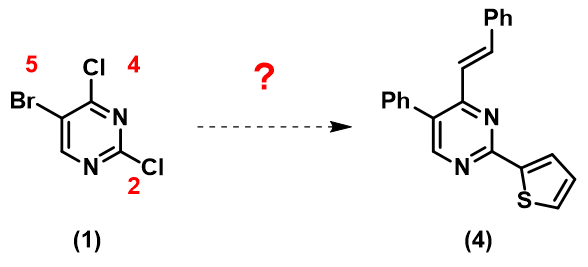
如果起始原料是多卤代的苯环,我们知道反应一般会遵循I > Br > Cl这个排序进行,但杂环体系中的多卤代底物就充满了不确定性。
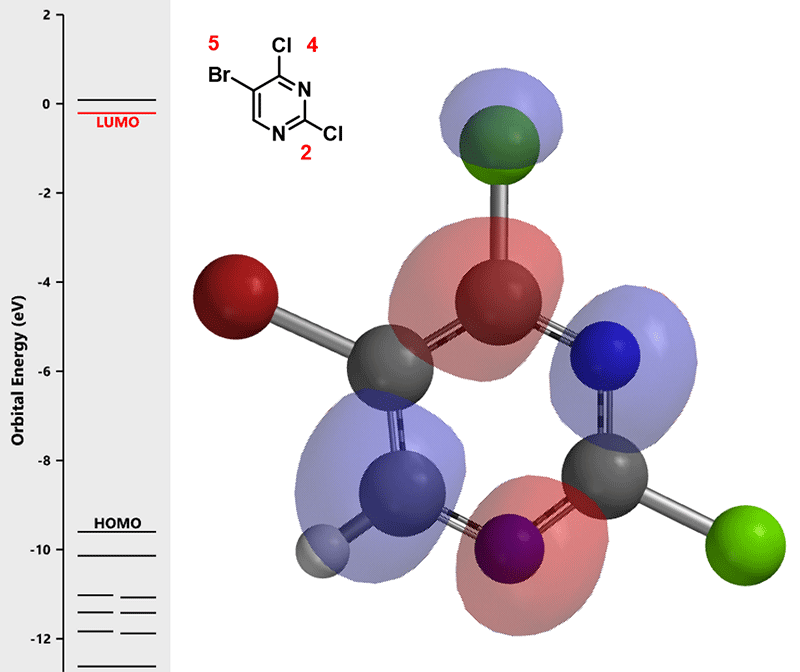
图一可知,LUMO图示意只有4-Cl的碳有很明显的LUMO lobe。因此,我们在钯催化下,并控制好反应温度在70°C,底物(1)可以与锡试剂A反应, 选择性生成C-4位烯基化的化合物(2):
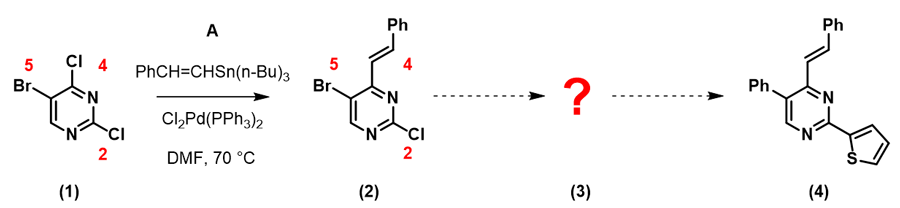
随后我们计算化合物(2)的LUMO:
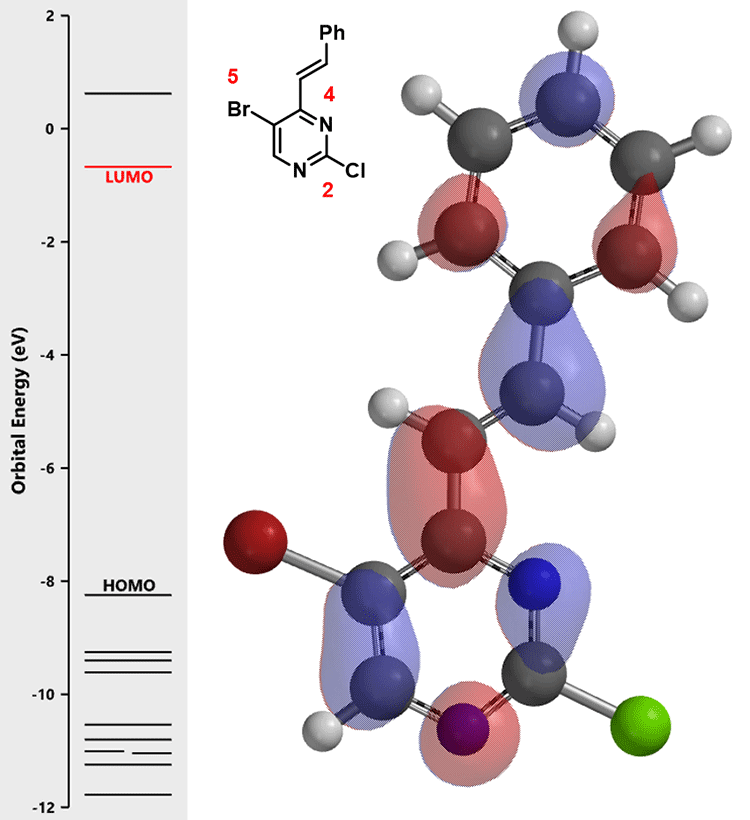
嘧啶2位和5位的碳均没有明显的LUMO lobes,我们进一步参考LUMO+1的轨道能级示意图:
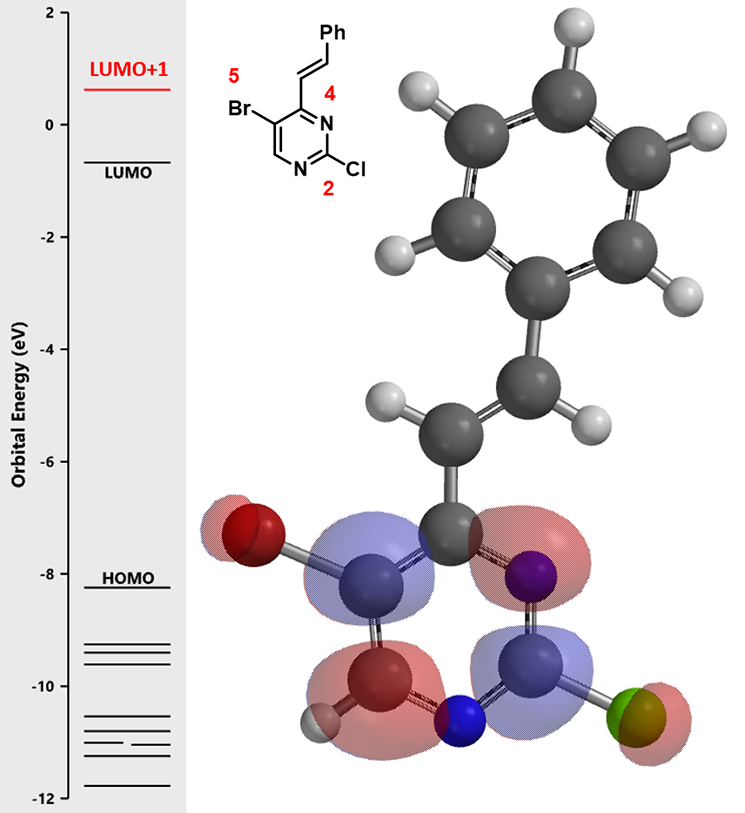
LUMO+1的示意图显示嘧啶LUMO lobes 在C-2位和C-5位上,但二者差别不明显。那么此时你是否认为我们要随机试验,撞大运了呢?答案是当然是NO! 山人自有妙计!
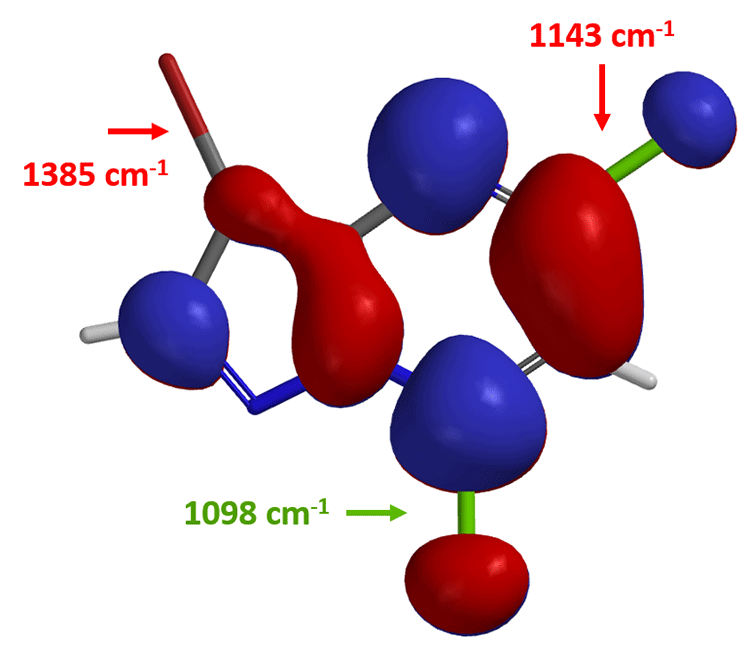
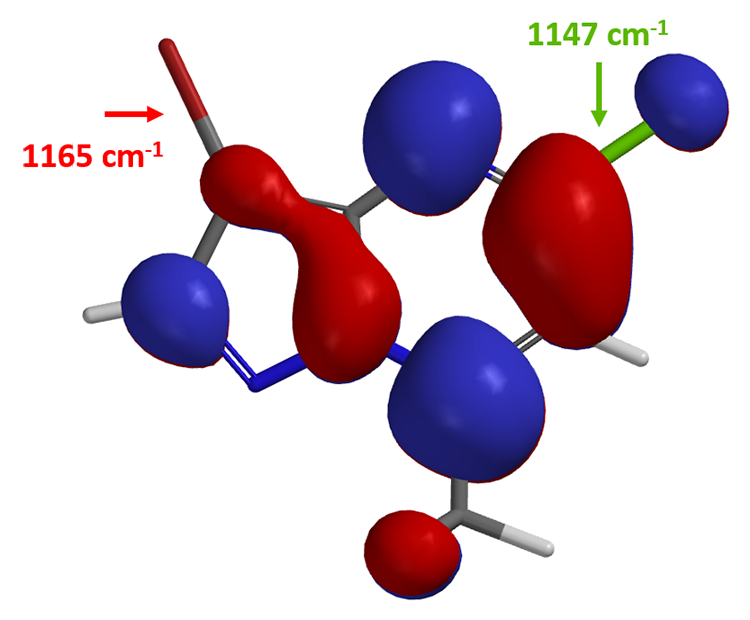
这里我们再介绍一个小技巧:计算C-X键的红外(IR)的伸缩振动频率来预测区域选择性。伸缩振动频率相对低的C-X键,该键键能越弱,越容易断裂。
我们打开计算工具,计算化合物(2)中嘧啶2位和5位C-Cl键和C-Br键的红外线伸缩振动频率:
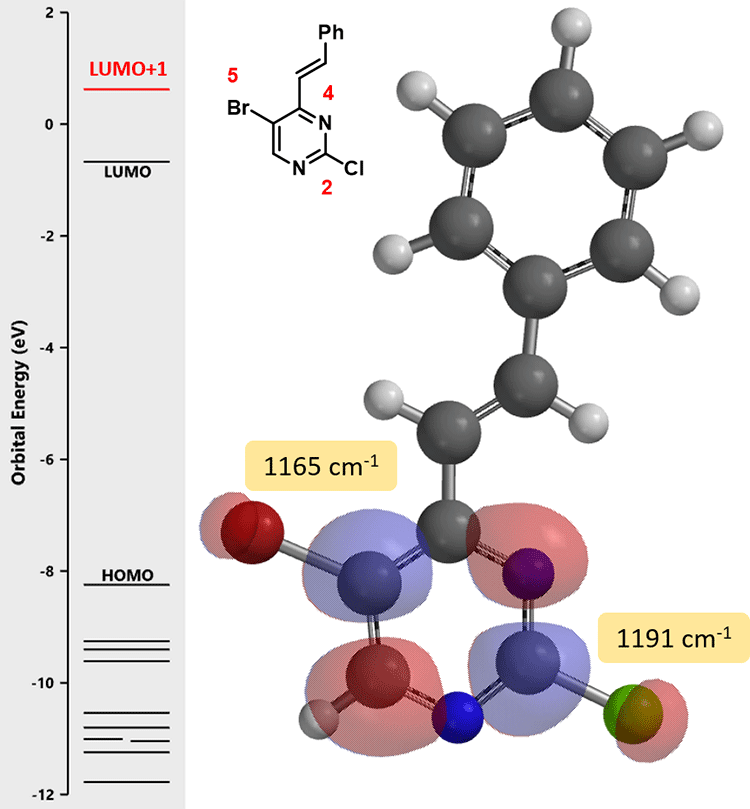
由此,我们可以很直观的做出比较: 5位C-Br键 (1165 cm-1) 比2位C-Cl 键(1191 cm-1) 的键能弱,更易发生氧化加成反应,实现嘧啶5位基团的引入。值得注意的是,起始原料(1)的最低未占轨道能量为 -0.21 eV,化合物(2)的LUMO+1能级的能量为0.62 eV,这就要求我们要提升一些反应体系的温度保证反应顺利进行。在相同催化体系下,升温至80°C,化合物(2)与锡试剂B反应生成产物(3)。

最后一步反应,化合物(3)的C-2位没有LUMO lobe,进一步考察LUMO+1,其轨道能量比化合物(1)和(2)更高些,约为0.70 eV。
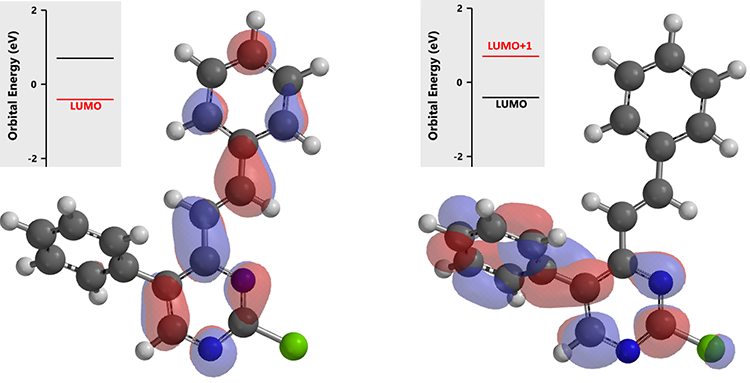
这就需要再进一步提高反应强度,升高体系温度至100 °C,可以成功得到C-2位芳基化的终产物(4)。
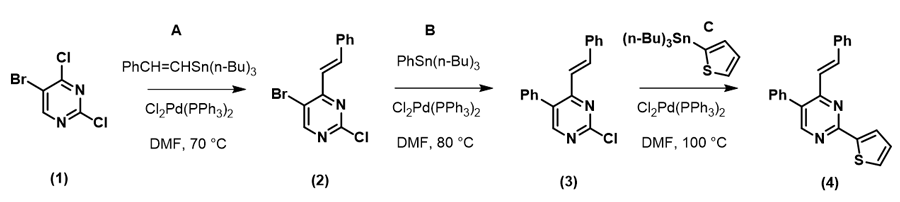
看完整个流程,您是否发现:我们可以尝试在不改变催化剂和溶剂等条件下,仅仅通过改变反应温度,不同阶段加入不同的定量锡试剂,中间步骤不经过纯化,通过一锅法实现三基团的顺序引入呢?
有没有超简单的感觉?提前计算一下,会比没思路的盲目尝试节省大量的时间和精力!
总结一下,通过QM计算我们优先考虑各反应位点LUMO 和 LUMO+1 lobes,如果不能明显判别,再继续计算红外线比较,通过评估各活性官能团参与反应的活性顺序,在一个相对普通的反应条件下, 设计出一条简洁且经济的合成路线。
本期我们也留了一个案例供大家思考:
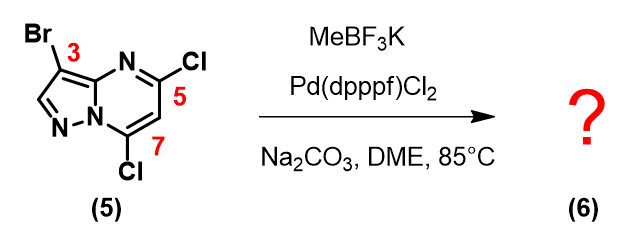
你能试着按上文所述的思路,参考如下所示的QM计算数据示意图给出一个合理的预测么?
引入一个取代基后,第二个取代基会在哪里发生反应呢?
温馨小提示:
之后的两节小课堂我们会继续介绍如何运用前几章学习的内容,介绍如何通过依次引入活性官能团继而引入不同化合物片段的应用以及对Chapter 1小试牛刀部分的一个答案详解!敬请期待!
本文由王秋月、石谷沁、郑重、王坚、卫小文编撰。
参考文献:
[1] Warren J. Hehre (2003). A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc.
[2] (a) J. Solberg & K. Undheim, Acta Chem. Scand. Ser. B. 1989, 43, 62. (b) J. A. Joule & K. Mills (2000), Heterocyclic Chemistry, Wiley-Blackwell, pp. 202-3.
[3] Richard M. Badger, J. Chem. Phys. 1934, 2, 128.
