












网站维护
系统内容更新/升级中


在第五章《QM在多卤素底物连续偶联反应中的应用》中,我们学习了如何利用QM计算LUMO/LUMO map和IR来预测多卤代杂环底物的钯催化反应活性顺序。在该章的“小试牛刀”部分,我们留下了一个问题,如下的芳杂并环化合物1 (图1),其反应位置选择性会怎样呢?
我们首先计算了化合物1的LUMO和IR,其LUMO lobe和三个碳卤键的伸缩振动频率如图2所示。
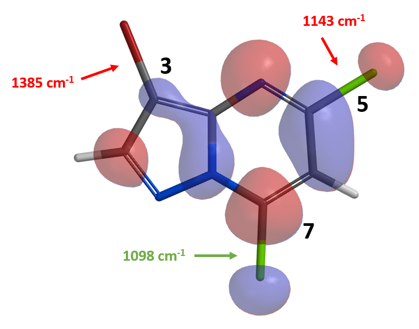
从图2中我们可以看出,连溴的C-3碳相对于连氯的C-5碳和C-7碳,碳原子上几乎没有LUMO lobe分布。IR计算可以得知,C-5位碳氯键的伸缩振动频率为1143 cm-1,C-7位碳氯键的伸缩振动频率为1098 cm-1。根据我们在第五章介绍过的规律,碳卤键的伸缩振动频率越低(波数越小),该键越容易断裂。而文献的实验结果也与我们的预测一致,和甲基氟硼酸钾反应成功的生成了C-7位甲基化的化合物2[1]。那么化合物2的偶联选择性是怎样的呢?让我们继续计算化合物2的LUMO和IR。
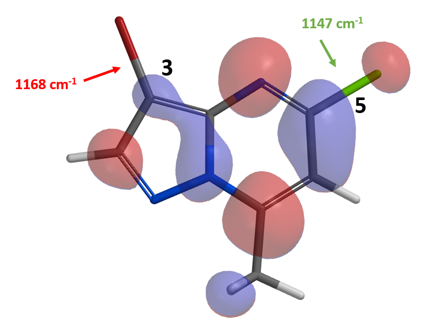
从化合物2的LUMO和IR计算结果 (图4) 可以看出,连溴的C-3碳和连氯的C-5碳上均存在LUMO lobe,C-3碳上的LUMO lobe还是相对较小。IR计算表明,C-3位碳溴键的伸缩振动频率为1168 cm-1,C-5位碳氯键的伸缩振动频率为1147 cm-1。LUMO和IR的结果均表明反应的优先位点是C-5位,同样和文献的报道相一致。在这里我们可以看到,芳杂并环化合物1和2的金属催化偶联反应的反应顺序与平常经验并不一致(X = I > Br ∼ OTf >> Cl >> F)。
从上一个例子中,我们得知在特定情况下,氯原子能够优先于溴原子发生反应,那么对于反应活性更高的含碘化合物,是否也会存在异于经验规律的情况呢?让我们来看两个例子。
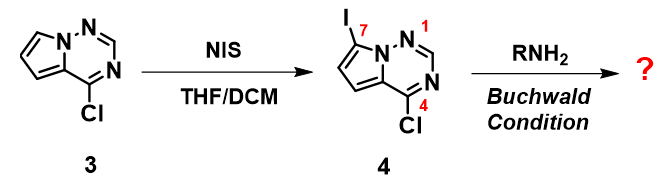
如图5所示,含碘化合物4能通过简单的碘代反应从化合物3制备。我们期望利用碘原子高度的反应性,进行钯催化的Buchwald反应得到C-7胺基化的产物。然而尝试了多种反应条件后,只能得到少量和连氯碳原子反应的副产物。那么这样的实验结果是否符合QM分析呢?我们随即计算了化合物4的LUMO和IR,见图6。
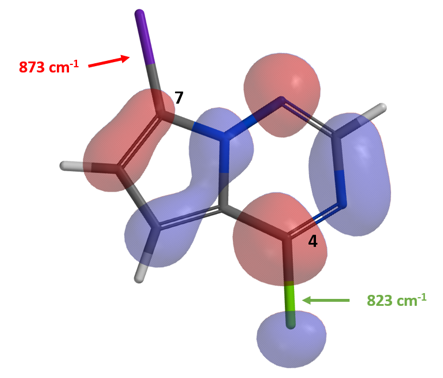
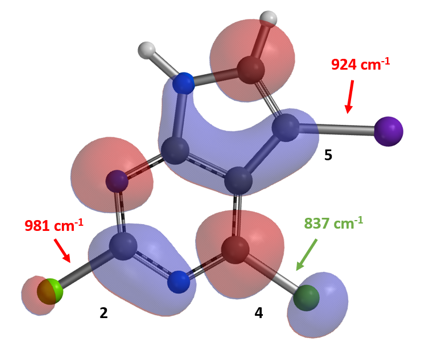
我们可以看出,连碘的C-7碳上的LUMO lobe相较连氯的C-4碳上的LUMO lobe更小,而C-4位碳氯键的伸缩振动频率也要小于C-7位碳碘键的伸缩振动频率。LUMO和IR的信息均表明,化合物4氯原子的反应活性高于碘原子,而这也和我们的实验结果相一致。这个例子告诉我们,在路线设计的过程中,利用LUMO等工具考察化合物性质是十分必要的,可以让我们避免一些弯路。
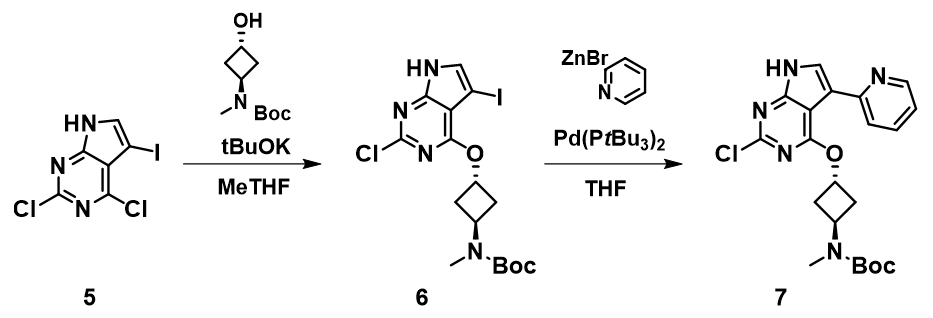
图7案例来自于美国辉瑞公司在OPRD发表的一篇论文[2],化合物5和仲醇化合物反应,选择性的得到了化合物6。由于发生的是芳香亲核反应,我们考察了化合物5的LUMO和IR,如图8所示,三个连卤碳上均有LUMO lobe存在,2位和4位的LUMO lobe大小差异不大, 但要显著大于5位的连碘碳。因此我们进一步考察了化合物5的IR,可以看到嘧啶4位的碳氯键具有最小的伸缩振动频率波数,应优先反应。实际实验得到的产物,也和QM的分析相一致。
在多卤素芳杂环底物中,金属催化偶联反应优先顺序不一定会遵循我们熟知的规律 (X = I > Br ∼ OTf >> Cl >> F),这点在双环体系中,缺电子芳环部分存在氯原子或溴原子、富电子芳环部分存在溴原子或碘原子时体现的尤为显著。这时,QM计算提供的LUMO和IR的信息就成为了有效的判断卤素反应活性的工具。我们需要在合成路线规划时通过综合QM计算得到的关键参数,确保正确判断反应位点,无需踏入被计算预测为不可行化学的弯路。
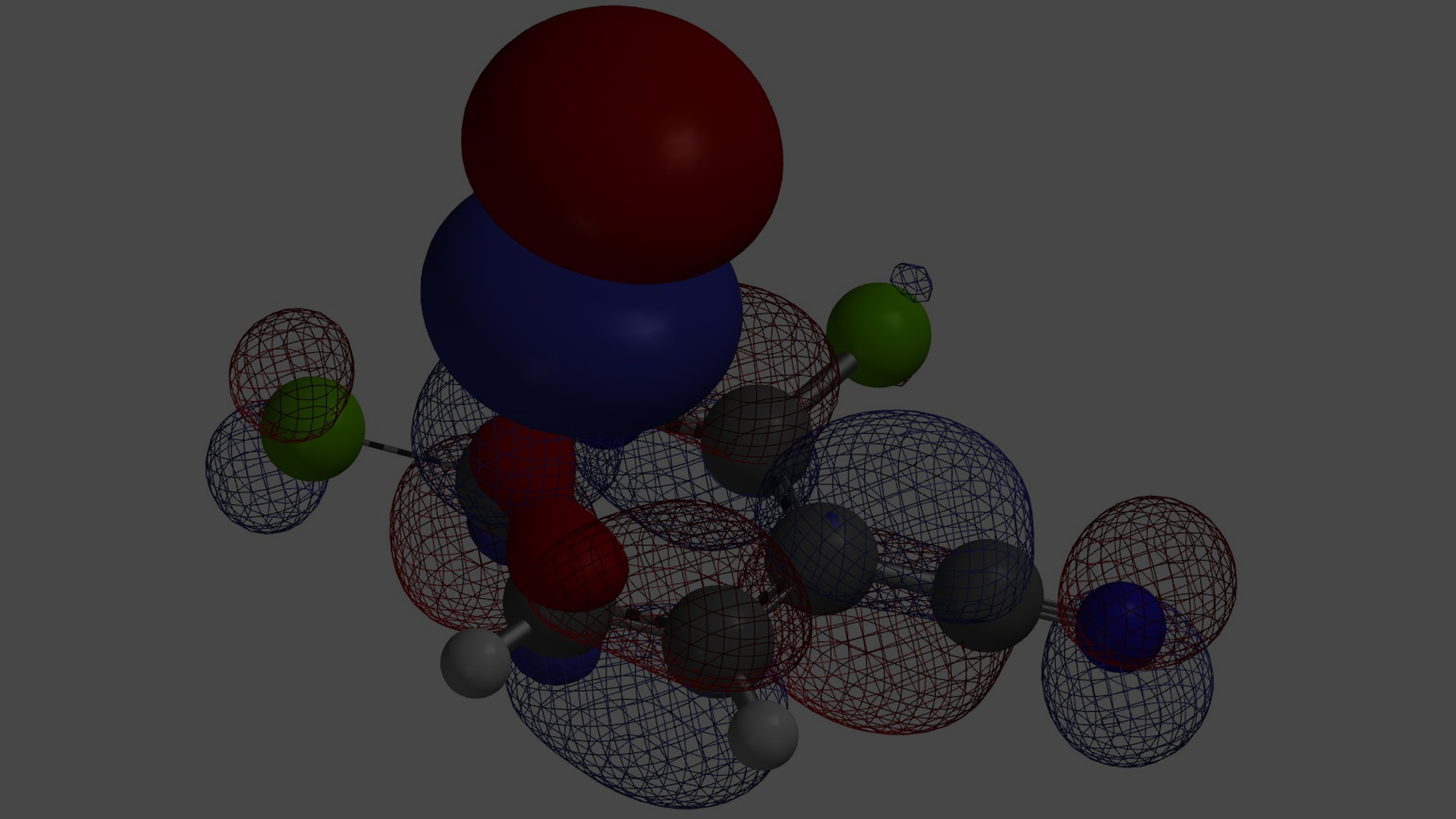
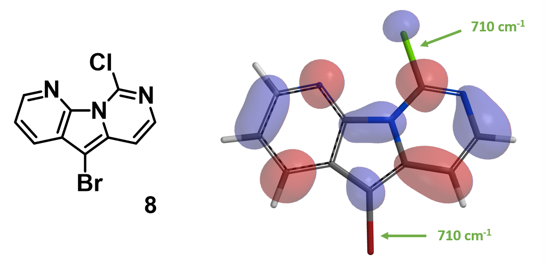
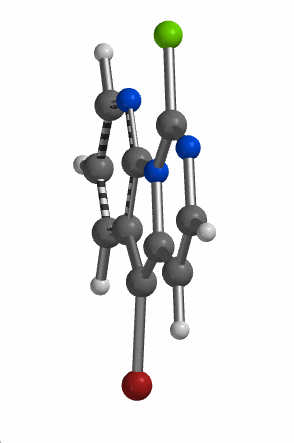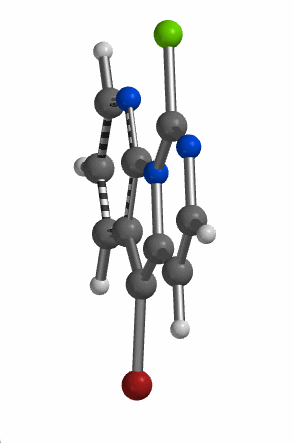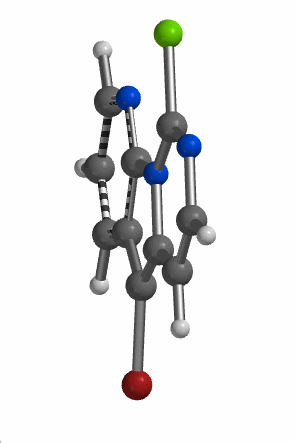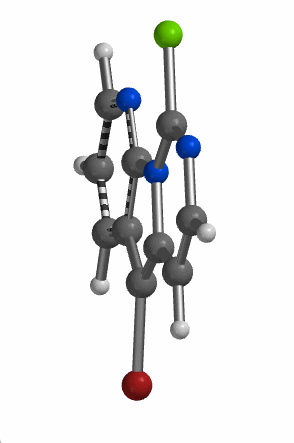
当考察的多卤代芳杂并环化合物从双环变成三环时,情况会不会有所改变呢?我们来看一下下面的5-溴-9-氯吡啶并吡咯嘧啶化合物,基于图9中的LUMO和红外信息,你能够猜到钯催化反应是优先发生在溴原子上还是氯原子上吗?控制反应选择性的是哪个因素呢?
本文由郑重、王秋月、卫小文编撰。
参考文献:
[1] X. Zusheng, L. Yangtong, Fused heterocyclic compound, preparation method therefor, pharmaceutical composition, and uses thereof. US 20160244432 A1, August 25, 2016.
[2] Y Tao, N. F. Keene, K. E. Wiglesworth, B. Sitter, J. C. McWilliams Org. Process Res. Dev. 2019, 23, 382.
