












网站维护
系统内容更新/升级中


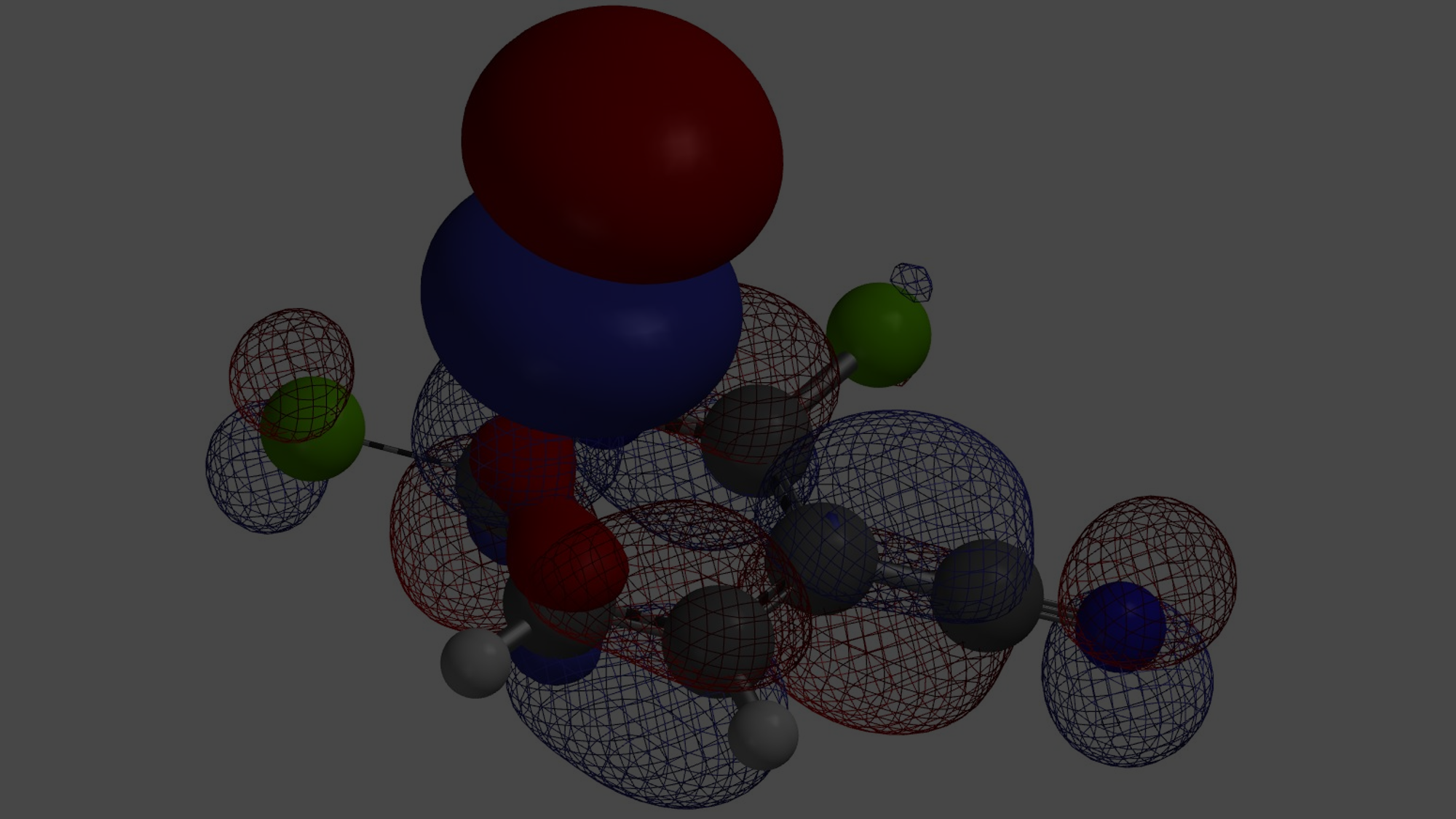
在第二章《亲电反应中HOMO计算的运用》的“小试牛刀”部分我们留下了一个问题:通过分析芳香底物D的HOMO和13C NMR,可以发现该化合物的C6和C7位的HOMO lobe大小以及13C的化学位移都很接近,没有显著差别。这种情况下做亲电溴代反应,大家可能会推测得到C6、C7位的溴代混合物,这也与我们传统理论中的定位效应相符合。但是实际情况是只得到了C7位的溴代产物,是不是让人大跌眼镜?那么怎么去解释这一结果呢?
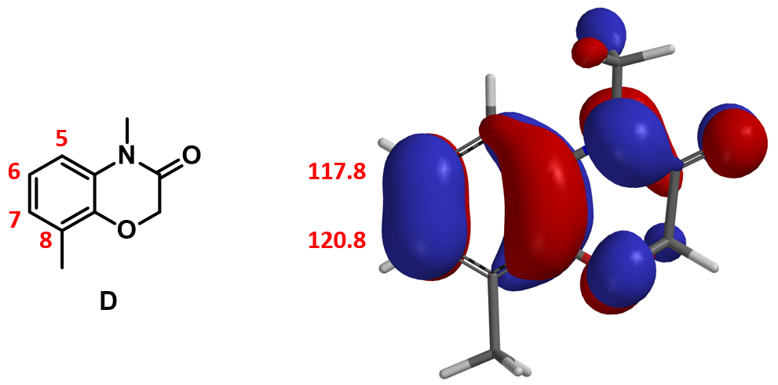
通过第九章《利用活化能计算预测吡唑氮烷基化的区域选择性》的学习,我们知道在HOMO与碳谱都无法有效区分反应位点的情况下,可以利用活化能计算辅助预测反应选择性,那么就让我们在这个案例中也尝试一下吧。
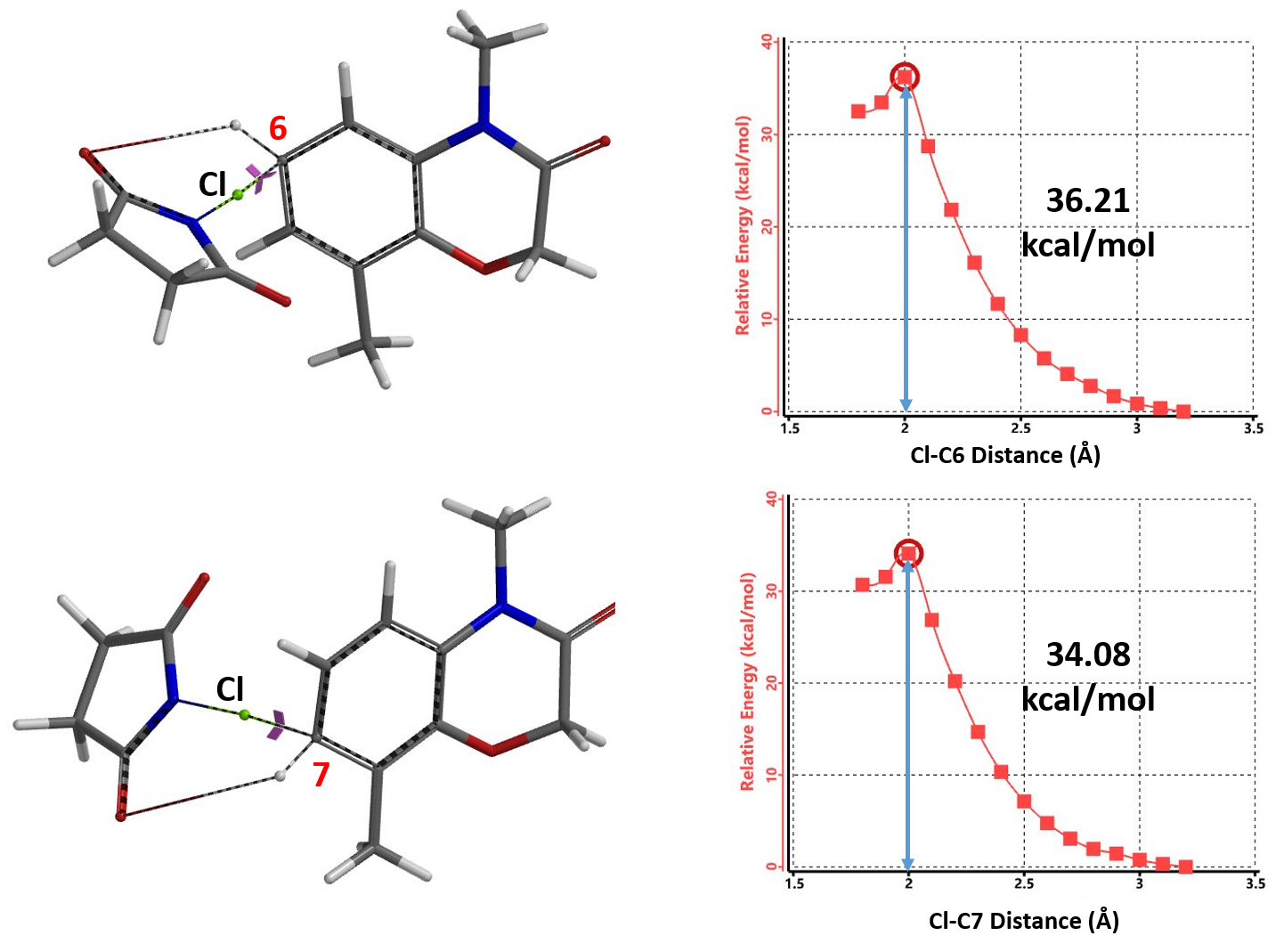
为了节省计算时间,我们用NCS代替NBS,分别计算了C6、C7位卤代反应的活化能。我们发现C6位发生卤代反应所需要的活化能约为36.21 kcal/mol,而C7位发生卤代反应的活化能约为34.08 kcal/mol(图2),C7位卤代所需要的活化能比C6位所需要的活化能要低,说明反应更容易发生在C7位。
我们还可以通过计算活化能差值,得到两个产物的大概比例。根据图3的阿伦尼乌斯公式,由于C7位卤代与C6位卤代的活化能相差2.13 kcal/mol,可以计算出C7位和C6位卤代产物比例约为113 : 1,这也就很好地解释了为什么只得到C7位的产物。
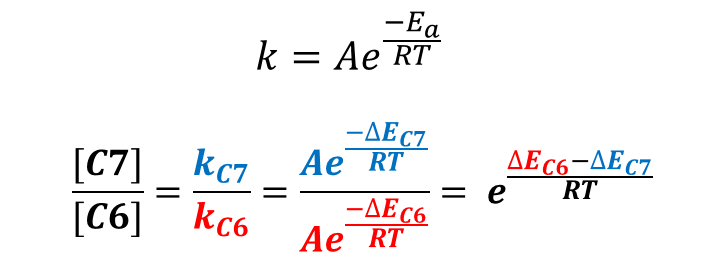
一般来说,活化能计算要求我们对反应机理理解较为深入,建立准确的反应模型的要求更高,计算时间也比较长[2,3]。那么有没有更方便、更省时的方法用于准确预测这类反应的区域选择性呢?
2018年,Jørgensen 等人在 Chem Sci 上报道开发了 RegioSQM 软件[1],用以快速预测芳环亲电取代反应的区域选择性。RegioSQM 的工作原理是先将芳环上的碳氢进行质子化,再利用PM3半经验方法计算出拥有最低自由能的碳(溶剂环境为氯仿),从而认为该碳原子是最具有亲核性的碳原子(图4)。不过在此案例中,利用 RegioSQM 来预测化合物 D 卤代的区域选择性时,也得出生成混合物的结论。
基于类似的原理,我们改进了 Jørgensen 等人的计算方式,利用精度更高的QM方法,使用DFT(密度泛函理论)中的ωB97X-D 泛函配合6-31G* 基组[4]计算相关的卤代正离子平衡构象以及相对能量,来预测最具有亲核性的位点。
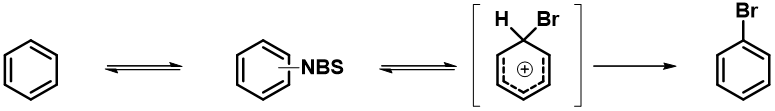
通过QM计算 C5、C6、C7这3个位点的溴代正离子的平衡构象,我们得到3个构象的相对能量(图5)。无论是在极性溶剂还是非极性溶剂的环境下,C7位溴代正离子的相对能量都是最低的,这说明C7位的溴代产物会是主要产物。第二种方法是不是更加直接、简单呢?
总结一下,在有扎实的反应机理基础时,我们可以通过反应活化能计算寻找过渡态,预测区域选择性以及反应产物比例,不过这类计算通常较为费时。作为替代,我们也可以计算、比较卤代正离子的相对能量,进行主产物预测,这种计算方法速度较快,也能获得可观的正确率。
本期我们再留一个案例供大家练习和思考:
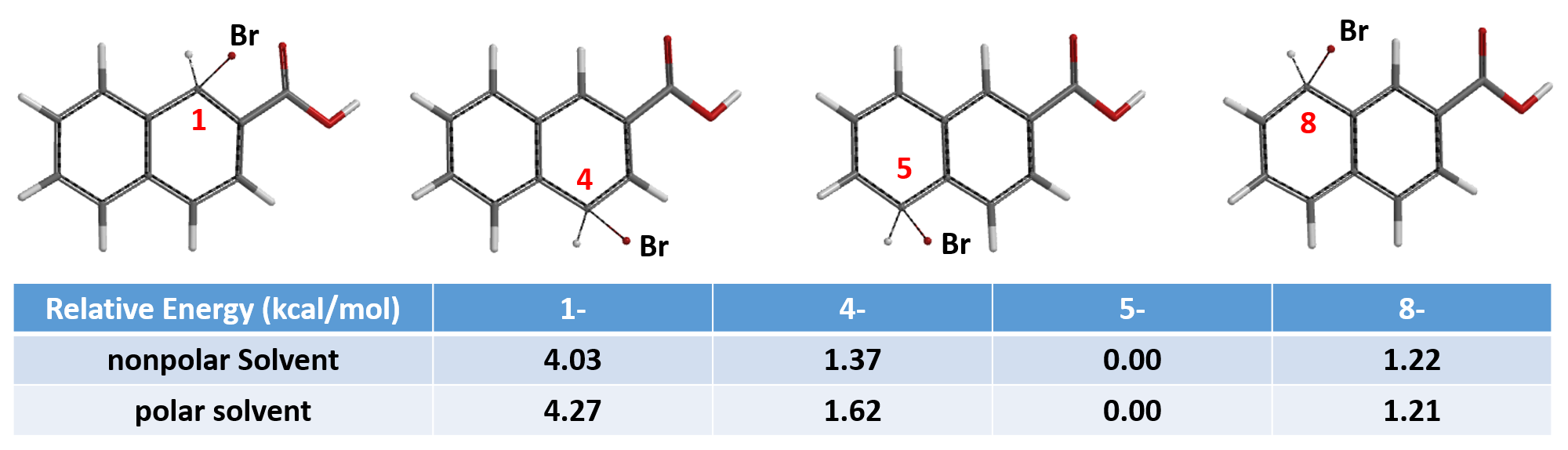
本文由许州、石谷沁、卫小文编撰。
参考文献:
[1] Jørgensen, M. et al, Chem. Sci., 2018, 9, 660. RegioSQM http://www.regiosqm.org
[2] Hoffmann, R., Malrieu, J.P. Angew Chem Int Ed Engl., 2020, 59, 12590 (Simulation vs. Understanding)
[3] 卤代反应中的过渡态结构与卤代试剂相关,在未来的章节中我们会继续探讨这一问题。
[4] Spartan’18 Tutorial and User’s Guide (2019). Irvine, CA, USA: Wavefunction, Inc.
