












网站维护
系统内容更新/升级中


从上世纪60年代初发现mRNA分子,到1998年首款反义寡核苷酸(ASO)药物获批上市,再到2006年RNAi技术获得诺贝尔奖,核酸药物疗法目前已经发展为基因治疗领域最具突破性的疗法之一。核酸药物能够从基因层面调控疾病相关的靶点水平或活性,具有靶点范围广,可编程,相对安全,迭代快等优势,使其成为生物医药研发的热门赛道,备受学术界和工业界的关注。
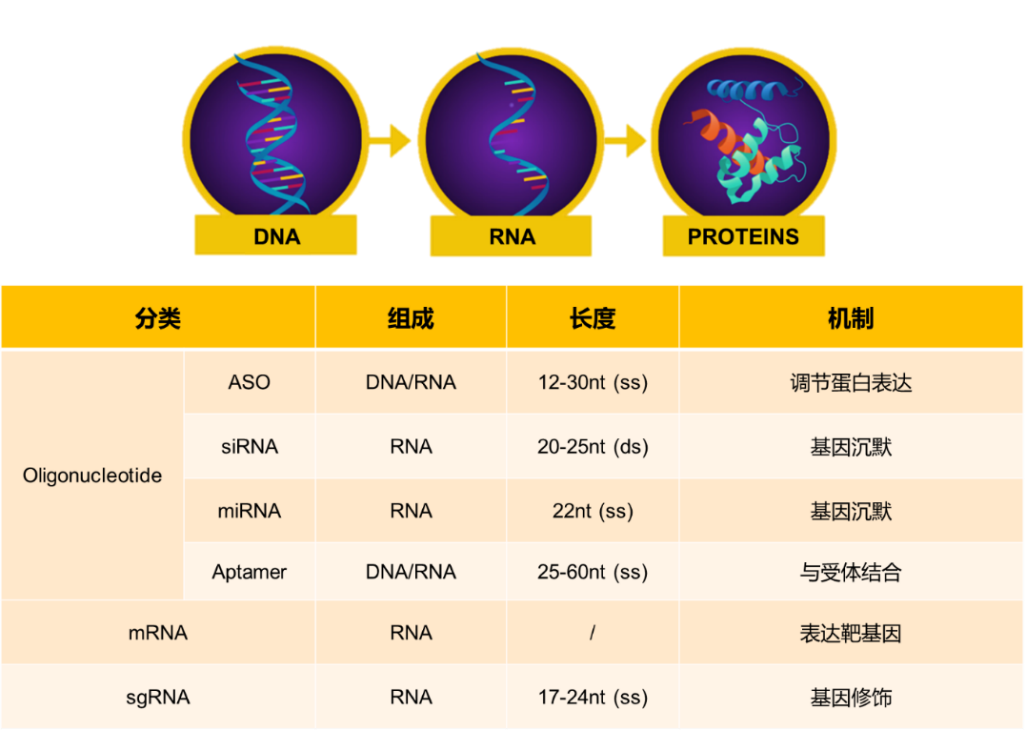
核酸作为生命体遗传信息的载体,分为脱氧核糖核酸DNA和核糖核酸RNA。根据中心法则,遗传信息从DNA传递到RNA,RNA通过转录和翻译生成蛋白质。然而,人类基因组中仅1.5%的序列能够编码蛋白质,蛋白质中仅10-14%具有小分子活性结合位点,这些蛋白被认为可以用作传统小分子药物与抗体药的“可成药靶点”。而对于数量庞大的“不可成药靶点”,开发核酸药物是其中一项重要的策略。与重组蛋白或抗体不同,核酸药物利用靶细胞内天然存在的转录和翻译机制发挥作用,无需复杂的蛋白质工程生产,具有设计简便,研发周期短,候选靶点丰富等优势。
根据作用靶点,核酸药物可以分为DNA靶向药物和RNA靶向药物,其中,DNA靶向药物需要进入细胞核发挥作用,存在与基因组整合风险。根据结构和作用机制,核酸药物可以细分为:1. 寡核苷酸药物:通过碱基互补或基因沉默的方式来调节基因表达的反义寡核苷酸(ASO)、小抑制RNA (siRNA)、microRNA (miRNA)或是利用单链寡核苷酸折叠形成的三级结构与靶蛋白特性结合发挥调节作用的核酸适配体(aptamer);2. mRNA 药物:直接表达靶点蛋白作为药物;3. 以sgRNA为代表的药物:修正因突变造成的错误,表达正常的靶点蛋白(图1)。
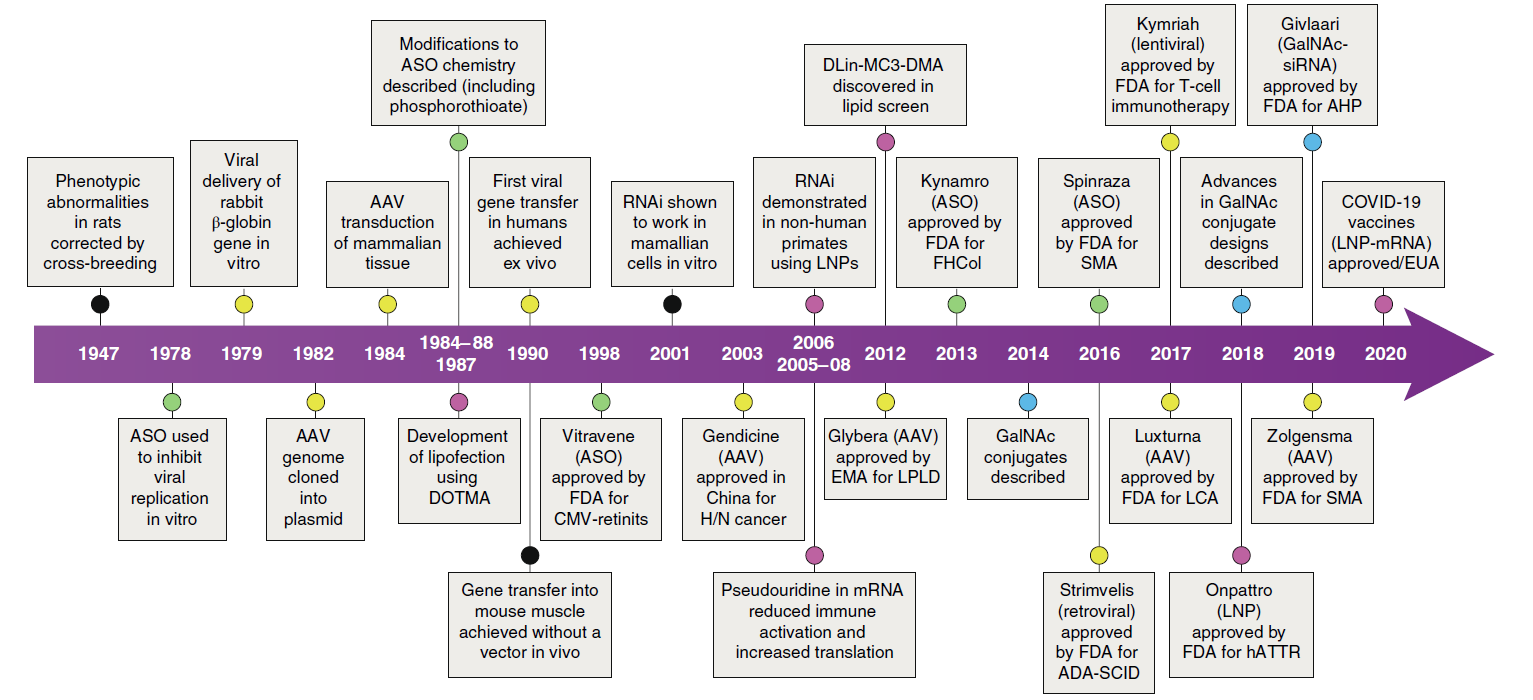
核酸药物的开发历史可以追溯到1947年,科学家发现在大鼠体内可以通过杂交的方式去纠正一些表型的异常,提示在基因水平的调控可以达到治疗疾病的目的,揭开了核酸药物开发的大幕。核酸药物的开发主要围绕两条主线进行:第一条是去寻找可以操纵或调控基因的有效物质成分;第二条则是探索把基因调控物质高效递送到靶向细胞的递送方式(图2)。
针对第一条主线,1978年Zamecnik和Stephenso发现了ASO,1988年首款ASO药物Vitravene成为最早的基于寡核苷酸的治疗方法。同年,Andrew Fire和Craig Mello因为在线虫中发现了RNAi的作用机制而获得诺贝尔生理学奖,并在2001年Elbashir等人首次利用体外合成的siRNA在哺乳动物细胞中实现基因表达的调控。2020年,mRNA疫苗的出现为人类抗击新冠肺炎疫情提供了有效的防治措施,mRNA在其他疾病领域的研发也引起了业界的关注[1] 。
围绕第二条主线,腺相关病毒(AAV)载体疗法更是其中最重要的治疗技术之一,催生了以AAV为载体的针对不同疾病领域的基因治疗产品。2014年GalNAc递送系统的出现赋予了核酸药物在全球范围内的第二次生命,GalNAc能够显著提高药物的肝靶向性。其他递送系统如脂质纳米粒(Lipid nanoparticle, LNP)、高分子聚合物、外泌体等的研究也在如火如荼的进行当中[2]。
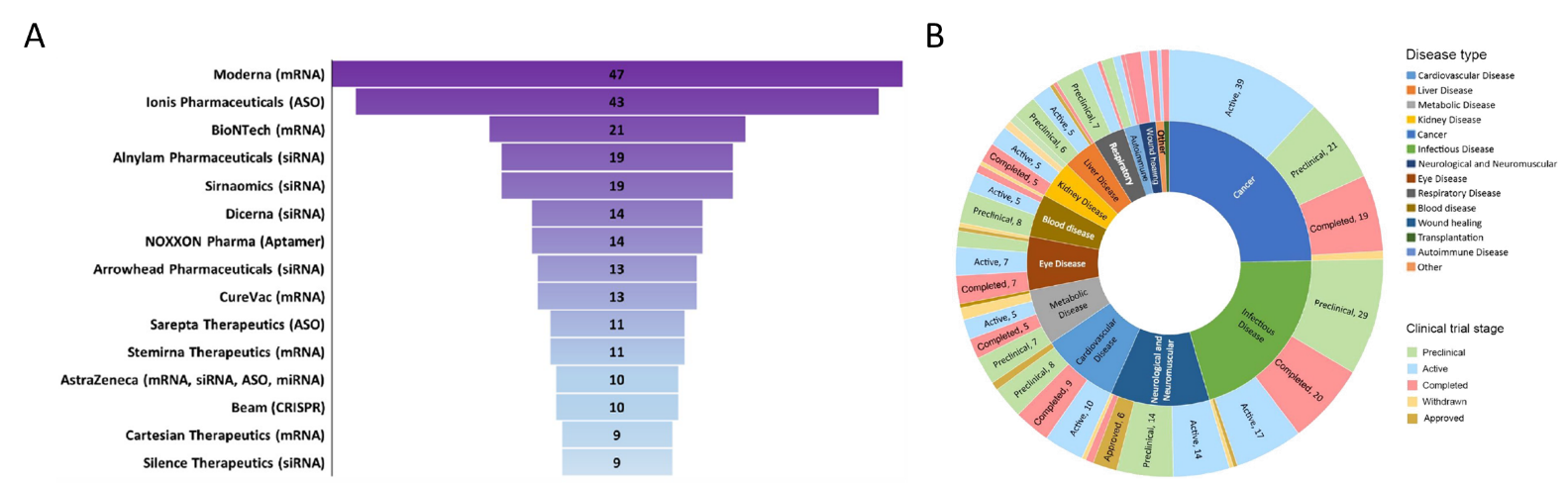
据弗若斯特沙利文(Frost & Sullivan)统计,小核酸药物全球市场规模从2016年0.1亿美元已增长至2021年32.5亿美元,全球小核酸药物临床在研管线类型包括ASO、siRNA、Aptamer、sgRNA、miRNA等。从医药公司全球在研的临床管线来看,在mRNA领域深耕的Moderna和BIoNTech分别有47、21条临床管线,专注于ASO和SiRNA领域的Ionis及Alnylam分别有43、19条临床管线(图3A)。如果从疾病领域来看,不论是临床前、临床试验进行中或完成阶段,核酸药物的研发以肿瘤疾病占比最多,消化道疾病、代谢性疾病等紧随其后,值得一提的是,神经及神经肌肉疾病等罕见病的核酸药物临床试验数量也在近年有所增加(图3B)。
核酸药物的临床前评价按照时间顺序,大致可以分为5个部分(图4),第一步,核酸药物的序列设计,根据核酸药物的分子类型需采用不同的设计策略,比如寡核苷酸药物采取的化学修饰及特异性筛选等方法,而mRNA药物采取的5’UTR & 3’UTR,核苷修饰,密码子优化等方法;第二步,根据设计序列合成相应的核酸分子,寡核苷酸通常采用固相合成的方法,mRNA则常采用体外转录方法;第三步,选择合适的载体将核酸包装,并高效、有效地在体内递送,常用的三类有以LNP为代表的纳米颗粒载体、以AAV为代表的病毒载体以及在核酸维度与核酸偶联增加器官靶向性的GalNAc载体;第四步,通过生物分析的方法验证核酸药物的递送效率,在细胞及动物水平上对药物的药代动力学进行评价,包括传统的液相检测LC-UV 及免疫因子及分子检测的ELISA、qPCR等方法;第五步,对到达靶器官的核酸药物进行概念验证,这是最重要的一步,包括对靶点调节验证,对药物体内、体外的药效及安全性进行评价,需要注意的是,核酸序列及核酸编码的蛋白序列是具有种属特异性的,需要通过非人灵长类动物(non-human primates,NHP)进一步进行免疫原性在内的安全性评价。
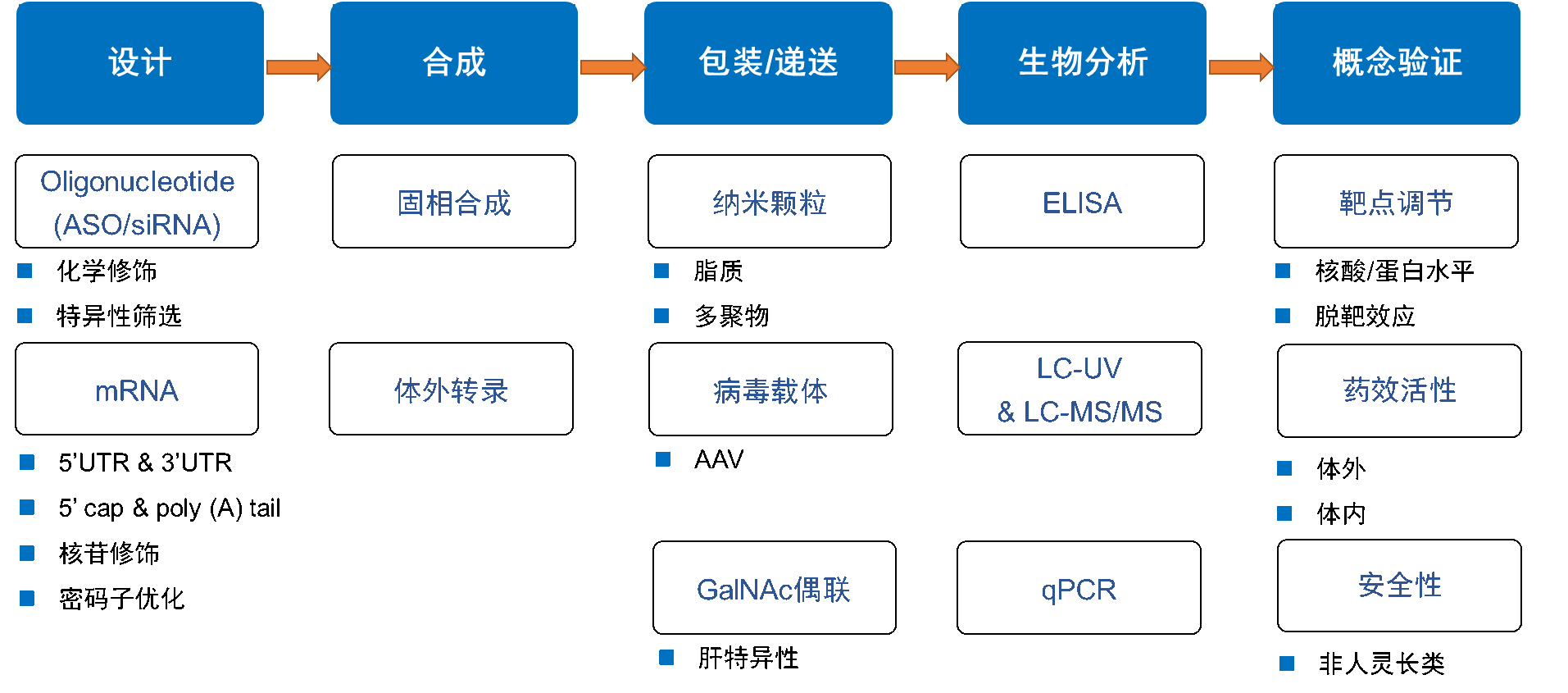
不同类型的核酸药物设计的侧重点是不一样的。针对ASO/siRNA药物的设计不仅需要强调特异性,还需要对核酸进行进一步的修饰,包括对核酸的骨架修饰增强稳定性,对碱基修饰增加亲和力。mRNA药物的序列较ASO/siRNA更长,包含了编码区域和非编码区域,对于非编码区域需要关注5’的帽子结构,Poly(A)的长度,5’/3’ UTR;对于编码区域,需要关注对密码子的优化促进mRNA能够准确高效的翻译成目的蛋白,同时需要关注碱基的修饰避免mRNA被宿主免疫系统清除,增强mRNA稳定性。现有的生物信息学、高通量测序等新技术发展,进一步提高了核酸药物设计的靶点特异性(图5)。
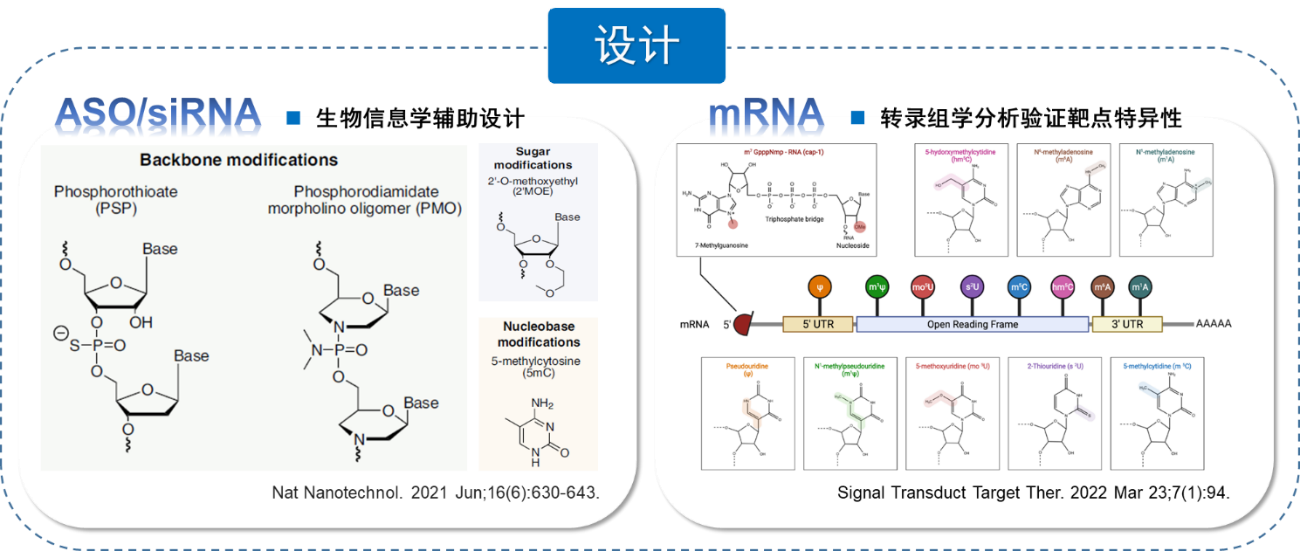
目前核酸药物的包装及递送方式常用的三种方式包括:1. 电转,对于一些不经任何包被处理的核酸药物通过物理的办法将其导入到目标细胞,图6实例中通过电转将编码GFP-mRNA电转入K562细胞,荧光强度表征电转效率为98%左右;2. 纳米颗粒递送系统,图6实例中将携带Luciferase荧光素酶报告基因的mRNA通过多聚物纳米颗粒包被后,注射进小鼠体内,通过动物活体成像检测Luciferase的荧光信号判断mRNA进入体内的效率及编码蛋白的效率;3. 腺相关病毒(AAV)载体系统,不同亚型AAV具有不同的器官靶向性,图6实例中将AAV病毒载体装载核酸药物在小鼠罕见病模型中,特异性靶向肝脏递送,能够看到mRNA在肝脏中的高表达,因为AAV携带GFP荧光表达,也能通过观测荧光表征判断药物在肝内递送的效率。
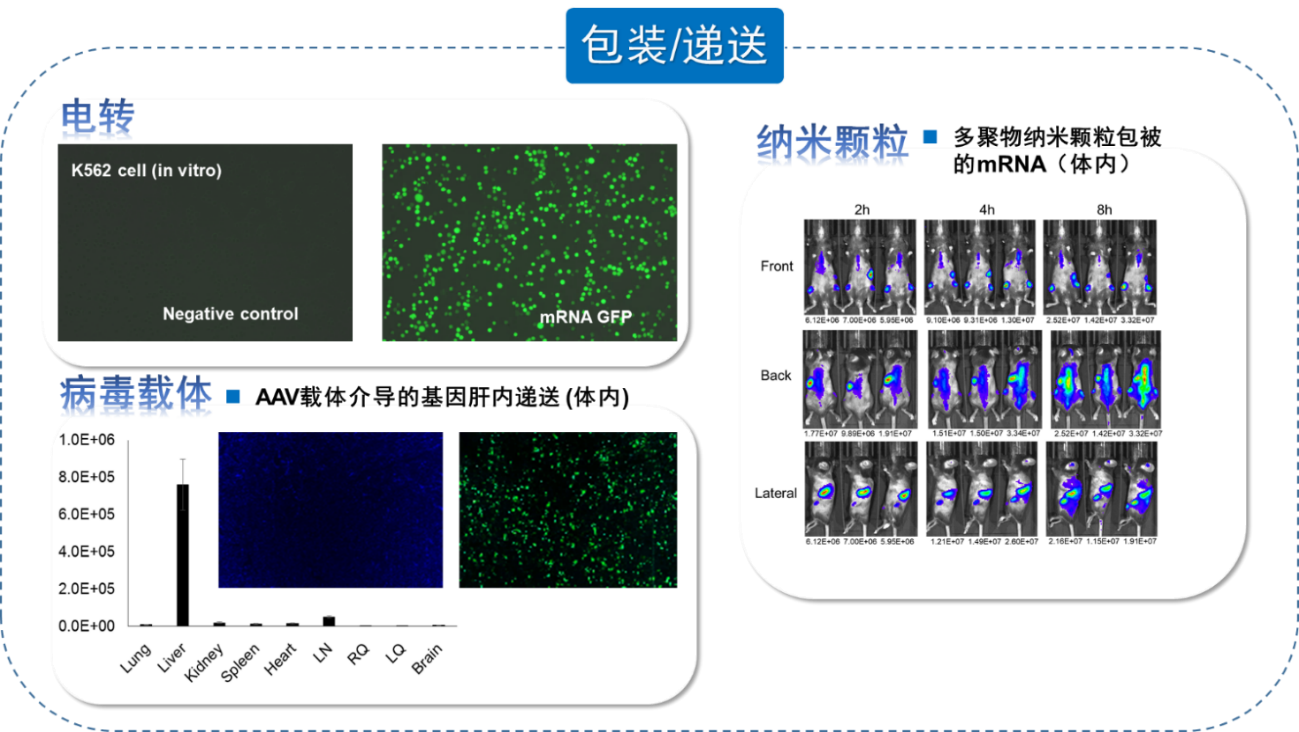
验证核酸药物递送效率的生物分析方法包括传统的液相检测及对细胞因子等分子检测方法,不同方法的敏感性、差异性及局限性不同,需要根据核酸药物的类型及观测指标和目的,选择合适的生物分析方法,比如qPCR方法具有高敏感性及特异性,但是不适合代谢物的检测,ELISA方法更适合对代谢物进行检测(图7)。
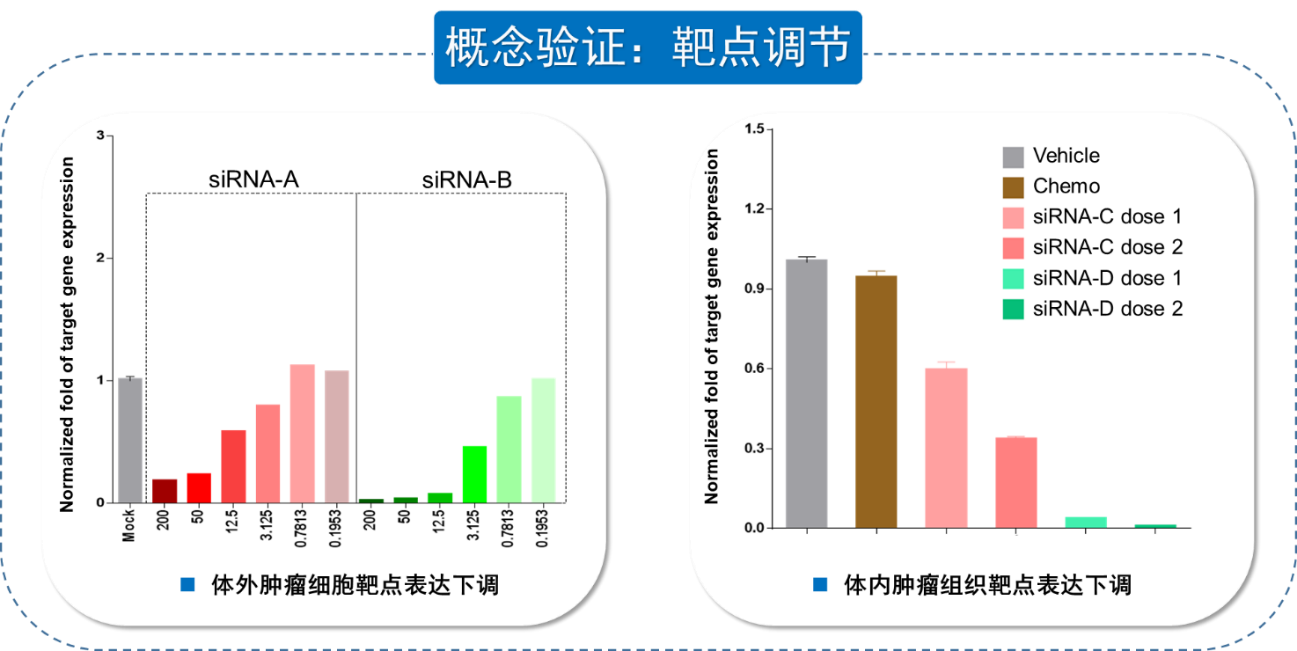
当足够量的药物能够抵达靶器官和靶细胞后,紧接着就是对核酸药物的概念验证。首先是对其能否有效调节目标靶点进行检测,图8以siRNA药物为例,在体外肿瘤细胞水平上检测两种siRNA药物的作用,结果显示二者都能浓度依赖性的下降靶基因的水平;在体内肿瘤模型上施用两种siRNA药物后,将肿瘤组织分离并进行靶基因的检测也得到相同的结果。以上体内和体外试验都证明了测试的siRNA药物能够有效降低靶基因水平,验证靶标被有效调节。
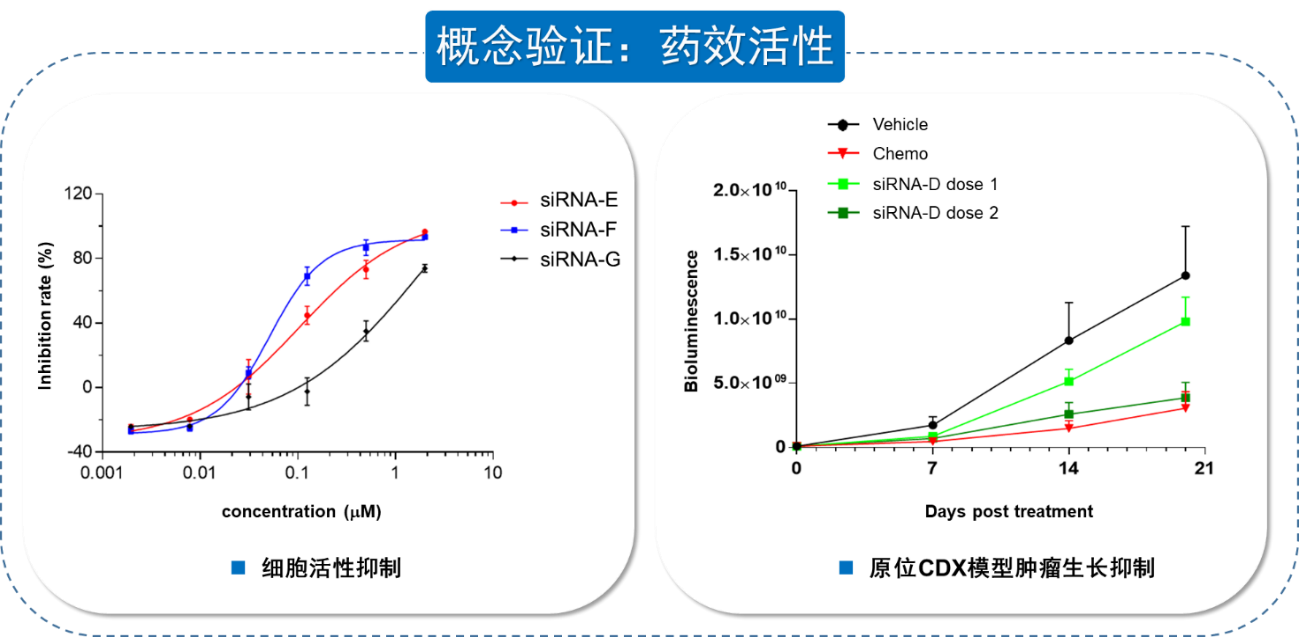
紧接着对靶点调节能否转化为药效活性进行检测,如图9,首先在体外细胞水平上用siRNA药物孵育,检测细胞活性,显示3种药物均成剂量依赖性的抑制细胞活性;在体内原位CDX肿瘤模型上测试siRNA药物的疗效,因为肿瘤细胞本身带有Luciferase荧光标记,通过Bioluminescence(生物发光)的强度表征肿瘤的进展,与化疗药(阳性对照组)、载体(阴性对照组)对比,siRNA药物能够剂量依赖性的抑制肿瘤生长,验证了药物对靶标的调节有效的转化为药效活性。
安全性评价也是核酸药物开发的重心,尤其是因为核酸序列及核酸编码的蛋白序列具有种属特异性,常常需要通过NHP进行免疫原性在内的安全性评价,包括在不同的时间点对施用药物后动物的血液采集,进行血液全细胞计数和血生化分析来评价药物的安全性。核酸药物的免疫原性主要分为两个方面:一方面核酸药物本身或其编码产物引起的;另一方面是核酸药物的病毒载体或者纳米颗粒载体引起的。针对载体可以通过检测抗药抗体(Anti-drug antibody, ADA)进行免疫原性评价。比如,如果选用LNP作为载体的药物,LNP载体中常含有PEG组分来延长药物的半衰期,可以通过抗PEG抗体来检测引起的免疫原性,而通过基于MSD的细胞因子检测则可进一步更全面的对药物免疫原性进行评价。
下文将通过两个案例进一步展示核酸药物的临床前评价侧重点及药明康德核酸药物研发能力平台。
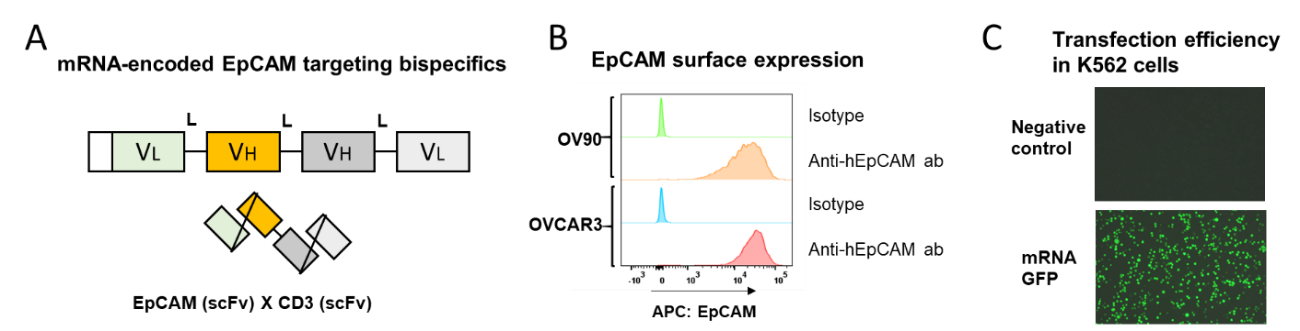
EpCAM-CD3双特异性抗体的作用机制是可以同时识别并结合两种不同抗原,同时与肿瘤表皮黏附因子EpCAM和T细胞表面CD3分子特异性结合,使得EpCAM高表达的肿瘤细胞被激活的T细胞杀伤。在本案例中,我们设计了一个能够编码EpCAM-CD3双特异性抗体的序列,不直接产生双抗,而是将设计的mRNA序列导入到肿瘤细胞内。首先是设计制造一个EpCAM x CD3抗体的mRNA序列并融合分泌肽(图10A),同时筛选细胞表面高表达EpCAM肿瘤细胞系OV90及OVCAR3,及低表达EpCAM的肿瘤细胞系Raji(图10B),紧接着采用电转的方法将设计的mRNA序列导入到生产细胞K562细胞中,通过观察GFP荧光强度判断电转效率(图10C),如果序列设计成功,能够在细胞中编码EpCAM-CD3双特异性抗体并分泌到上清液中。
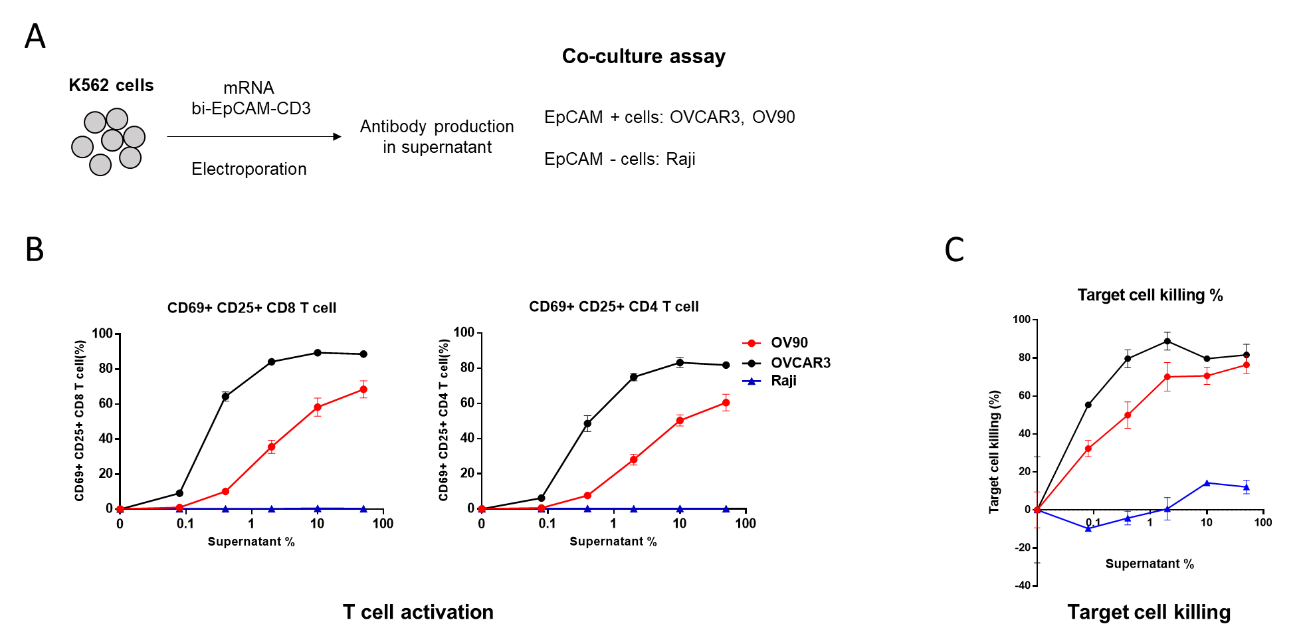
收集上清液加入到EpCAM阳性细胞或者EpCAM阴性细胞中与T细胞共培养,观察mRNA序列能否编码双抗并产生预期的药效(图11A),即产生的双抗能否介导T细胞的活化及活化的T细胞能否造成EpCAM细胞的死亡。结果显示,不论在CD8 positive的T细胞还是CD4 positive的T细胞,都只有在与EpCAM阳性的OVCAR3及OV90细胞及上清液存在的情况下才能被激活(图11B),同时通过靶向细胞实验也检测到被激活的T细胞能够杀伤EpCAM阳性的肿瘤细胞(图11C)。后续还需进行更深入的体内药效及安全性等试验的检测,本文中暂不详述。
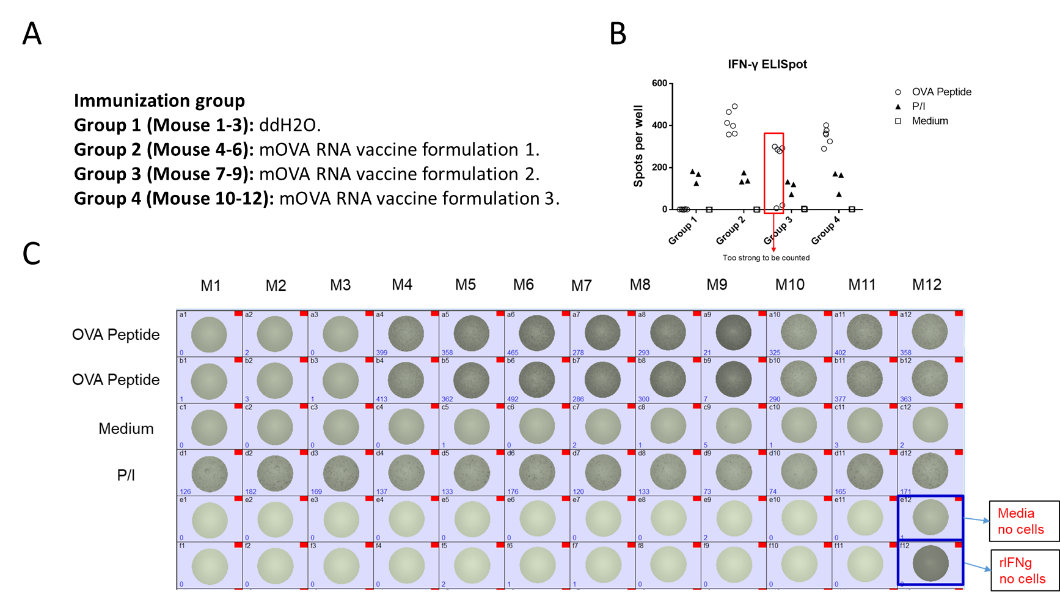
本案例中我们针对免疫学研究常用到的工具抗原—OVA抗原的mRNA疫苗,它能够编码OVA的肽段。我们将mRNA疫苗通过不同的载体和配方包被负载,对免疫健全的小鼠进行注射(图12A),随后将免疫后小鼠的脾脏分离出来,通过ELISPOT实验检测分泌的IFNg的脾脏细胞,只有在小鼠中能够成功编码产生OVA肽段并成功刺激小鼠的获得性免疫细胞激活的情况下,在体外用OVA肽段再次刺激才能够进一步活化并产生IFNg。结果显示不同配方的mOVA RNA 疫苗能够引起小鼠不同强度的免疫反应(图12B&C)。
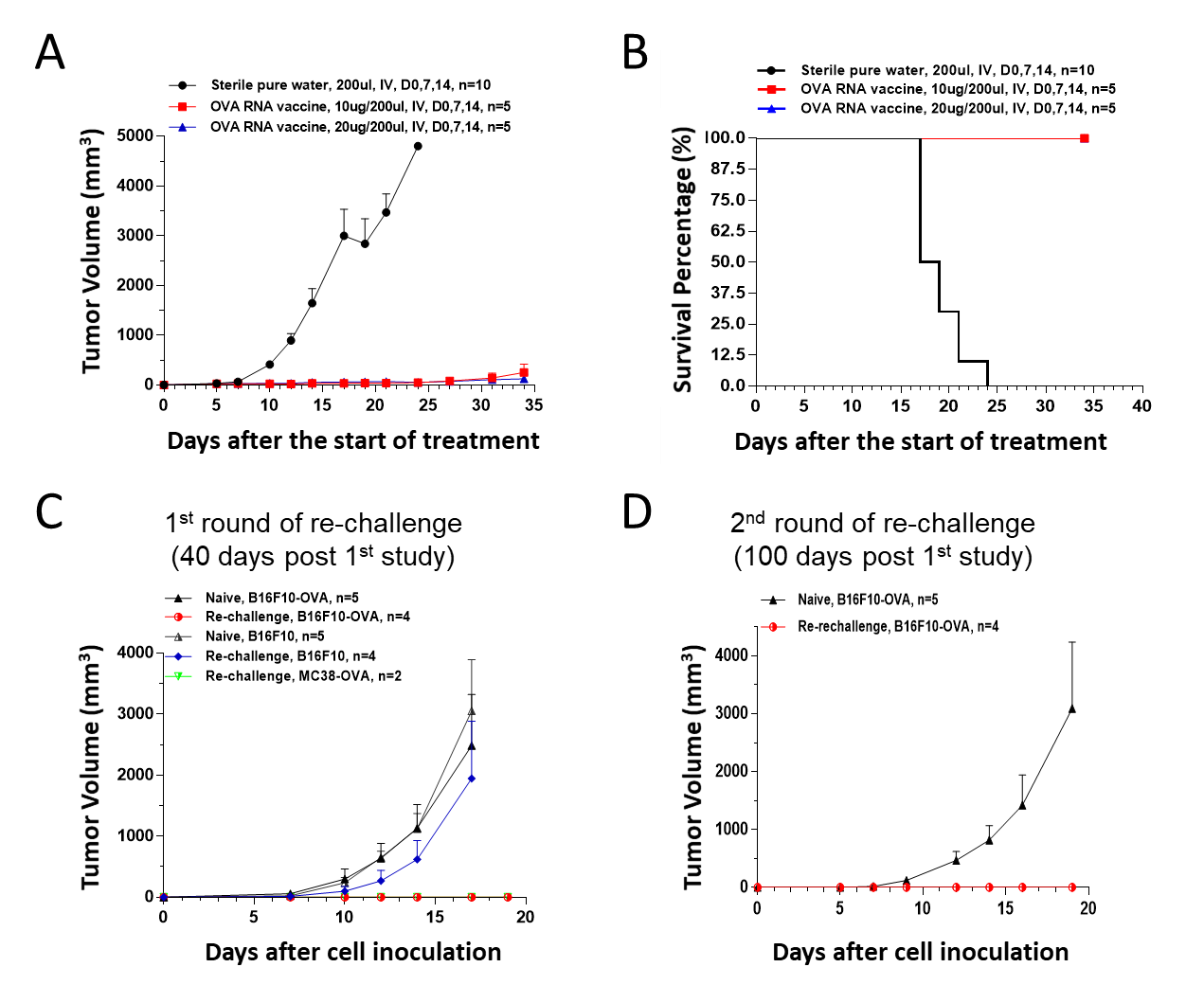
随后,我们在过表达OVA的B16F10-OVA小鼠模型中对OVA RNA疫苗能否提供抗肿瘤活性进行了检测,结果显示OVA RNA疫苗能够完全抑制B16F10 OVA小鼠肿瘤的生长(图13A),并且OVA RNA疫苗能够显著延长B16F10-OVA荷瘤小鼠的生存时间(图13B)。此外,在40天后我们对疫苗免疫存活后的小鼠分别用表达OVA和不表达OVA的肿瘤细胞进行再攻击,发现疫苗免疫后的小鼠能够有效抵抗表达OVA的肿瘤细胞,说明OVA RNA疫苗在小鼠体内诱导了OVA特异性的免疫记忆(图13C),在100天后疫苗免疫后的小鼠仍能够有效抵抗表达OVA的肿瘤细胞,说明疫苗诱导了持久的特异性免疫记忆。
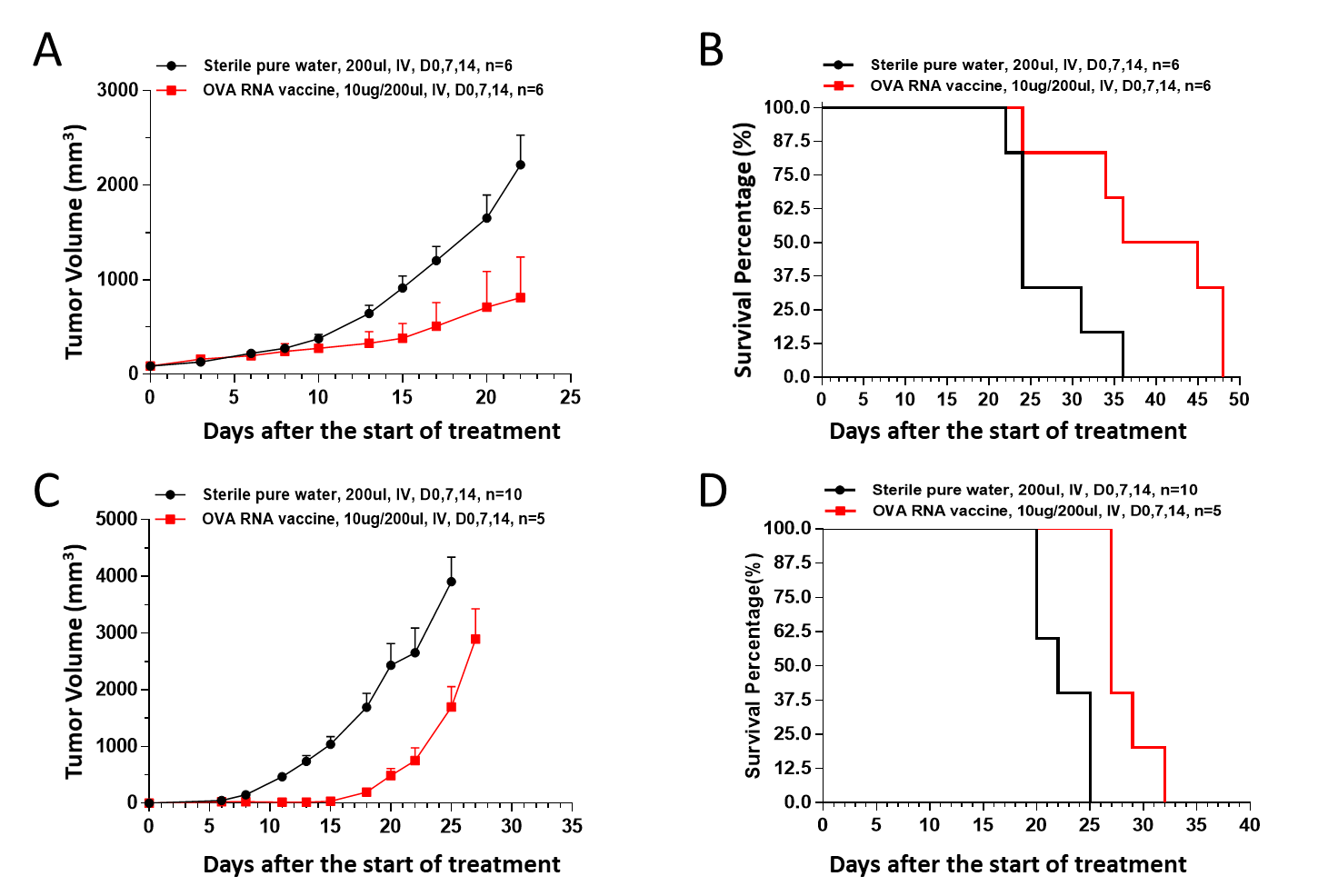
除了B16F10-OVA我们也在MC38-OVA(图14A&B)、EF.7-OVA(图14C&D)小鼠肿瘤模型上检测了OVA RNA疫苗的抗肿瘤药效,也得到了在B16F10-OVA小鼠模型上相似的结果,即OVA RNA疫苗能够显著抑制MC38-OVA和EF.7-OVA肿瘤的生长,并延长荷瘤小鼠的生存时间。
经过多年的发展,核酸药物有望成为继小分子药、抗体药物之后的第三大药物类型。其靶点范围广,相对安全,设计灵活等优势使核酸药物的适应症范围从罕见病扩展至慢性疾病及肿瘤领域,然而,核酸药物也面临着诸多挑战,如在体内的不稳定性,缺少精准的递送系统,潜在副作用等,这使得核酸药物的研发任重道远,未来仍然需要科学家们不断革新技术,扩展核酸药物的适应症,满足未满足的临床需求,从而造福更多病患。
参考文献:
[1] Nat Nanotechnol. 2021 Jun;16(6):630-643; Nat Rev Drug Discov. 2018 17, 261–279.
[2] Nat Rev Drug Discov. 2019 May;18(5):358-378.
[3] J Med Chem. 2022 May 26;65(10):6975-7015.
