












网站维护
系统内容更新/升级中



合成结构新颖的药物小分子一直是药物研发中的瓶颈。已建立好的合成路线也往往在药物分子优化的过程中需要不断调整,甚至是重新设计。
如何快速通过这个瓶颈?药明康德研究服务部的科学家们认为把量子化学计算结合到我们的有机合成路线设计中,可以预见风险,优化策略,提高合成的成功率。
量子化学的基础是薛定谔方程。由于它的计算复杂性,在二十世纪五十年代,化学家福井谦一提出了相对简化的前线轨道理论(Frontier Orbital Theory),用相对比较直观的形式将量子化学和有机化学结合起来,对化学反应做定性分析。
近几十年,量子力学中的密度泛函理论(Density Functional Theory)不断地发展改善, 计算机硬件的算力也快速增长,一般的化学计算早已融入进日常的合成工作。过去几年中,药明康德研究服务部(WuXi AppTec, Research Service Division)致力于将量子化学计算应用到有机合成化学领域,从中积累了很多宝贵的经验。科学家们因此更加坚定,进一步深化量子化学计算,结合人工智能路线设计,连接量子化学与机器学习,一起创造未来。
通过在跨国药企,世界知名学府,多国化学学会年会上的展讲,药明康德在这个领域的研究在业界也获得了广泛认可, 并于2020年,完成出版“Quantum Mechanics for Organic Chemists”的第一份合辑,多渠道同步发布中文版 “量子力学在有机化学上的应用”。
详情可见: ![]()
1. 反应可行性和底物的反应活性预测:对于某些反应,我们通过计算并结合实验数据,可以确定反应发生的能量阈值。基于此,我们可以对其他未知底物进行计算并预测其反应活性,从中筛选出可行性高的底物进行实验,节约大量人力物力和时间成本。
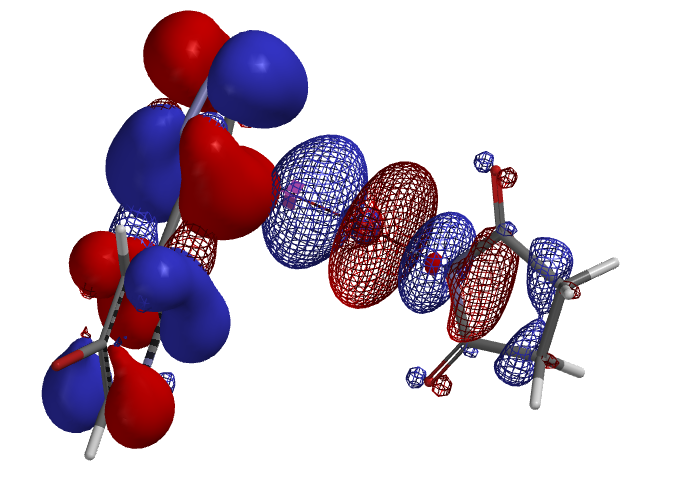
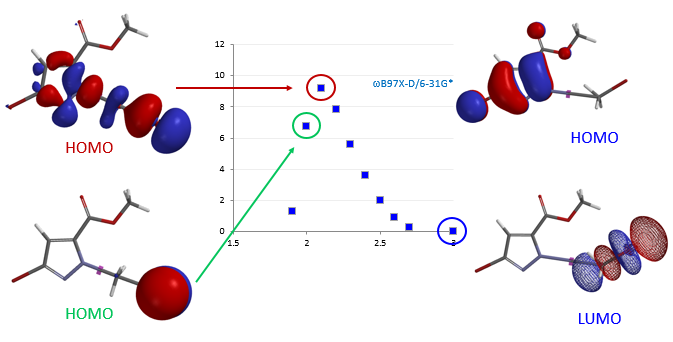
2. 亲核反应的可行性和区域选择性预测:对于亲核取代反应,我们通过对反应底物的计算结果分析(如HOMO/LUMO轨道,静电势图等)来预判反应的可行性,在底物中有多个反应位点存在的情况下,用计算来预测及解释亲核取代反应的区域选择性。
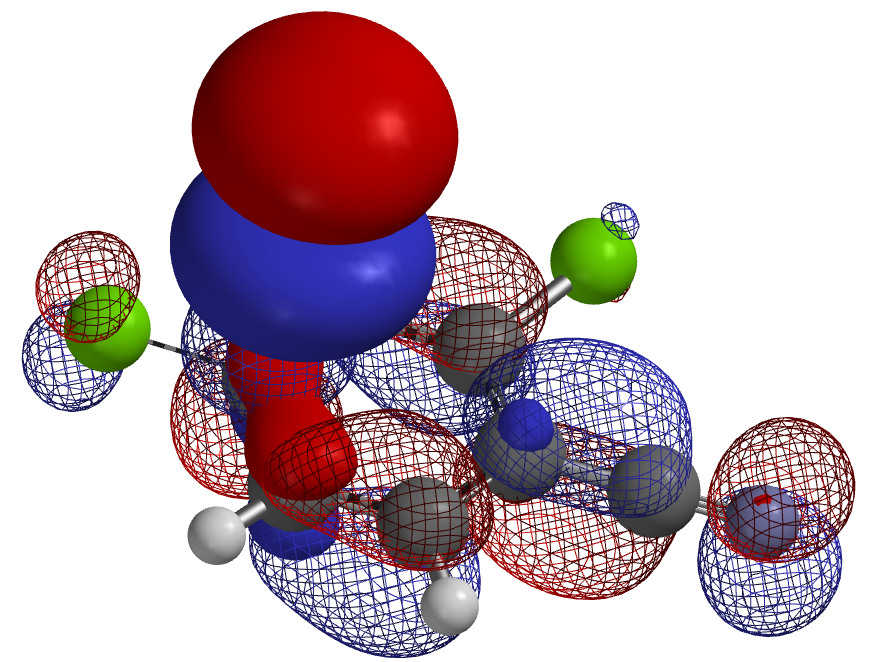
3. 芳香体系中亲电卤代反应的区域选择性预测:如何确定芳香化合物的亲电卤化的优先反应位点一直是个挑战,运用量子化学计算准确预测卤代反应位点的优先次序,高效地在芳环上引入取代基团。
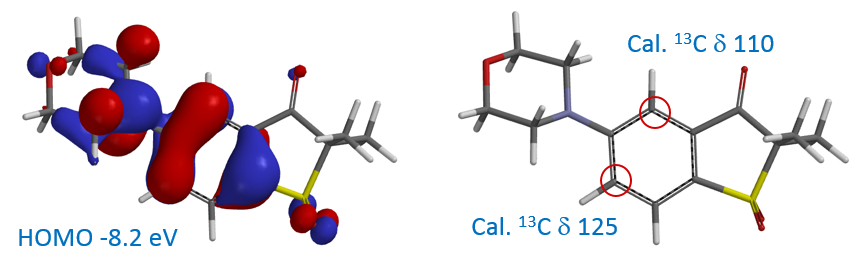
4. 判断金属催化偶联反应的区域选择性:在反应底物中有多个反应位点存在的情况下,通过计算反应底物的红外谱图,从中读取相关C-X键的伸缩振动频率,来判定优先发生反应的位点。
5. 预测特定反应的速度:对于一些难以通过上述手段进行预测的反应,我们可以通过计算反应的活化能,来预测反应的活性和速度,从而判断反应的可行性,区域选择性等等。
我们团队还在不断尝试开发新的应用。 如果你对这项研究感兴趣,可以扫描以下二维码关注IDSU微信公众号,收获第一手学习资料:

若有业务需求,可以联系我们了解更多信息。
