












网站维护
系统内容更新/升级中


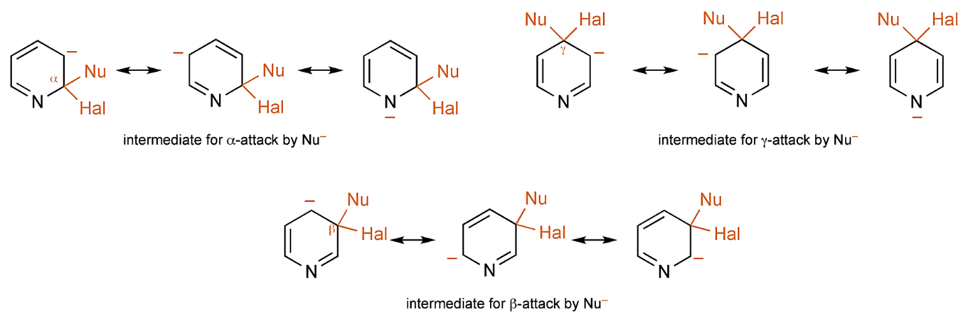
在第二十五章《评估反应活性的简单方法-LUMO和HOMO的能量差中》中,我们介绍了利用LUMO / HOMO的能级能量差来快速评估相关反应活性,并根据反应位点前线轨道 (FMO) 的可用性来选择合适的轨道,计算能级差。本章则将对芳香亲核取代反应中,轨道能量的高低与反应活化能之间的相关性进行考察分析。
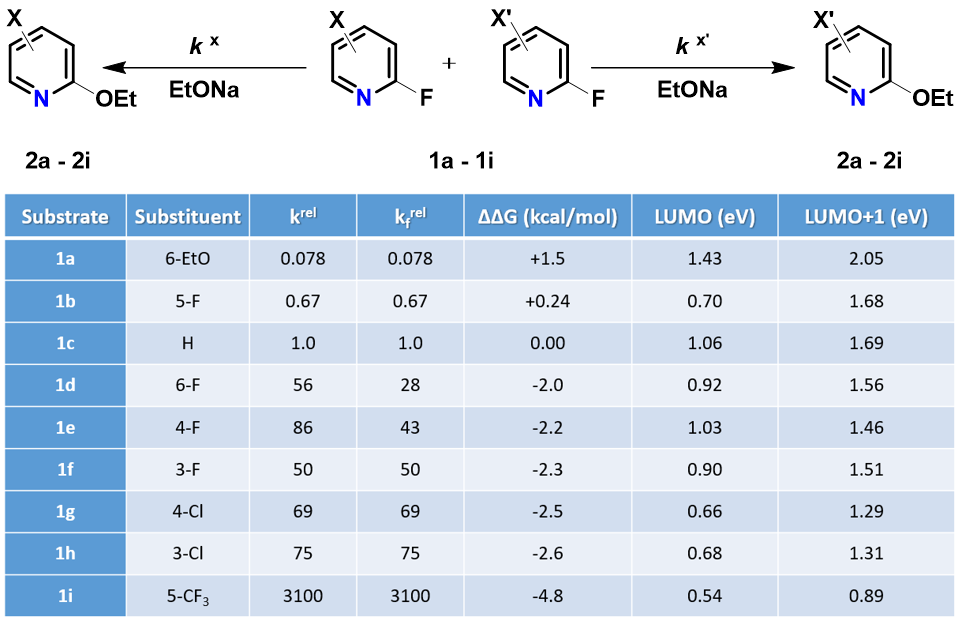
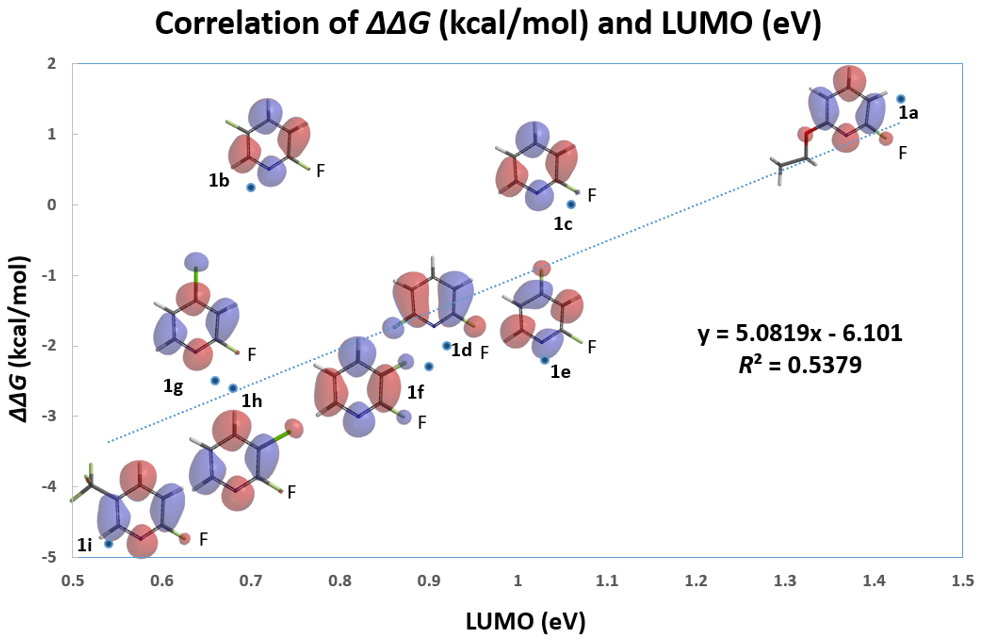
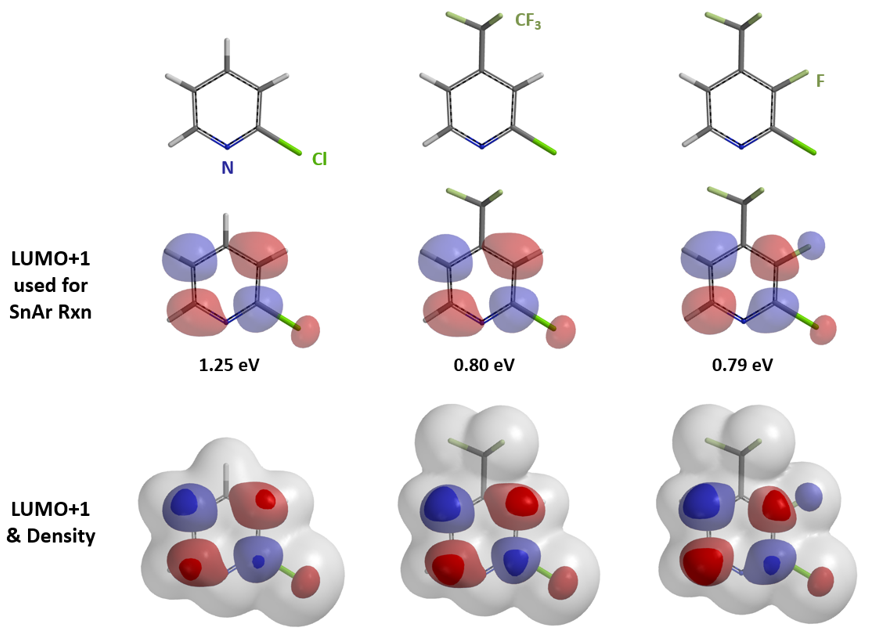
我们随即考察了取代2-氟吡啶原料的LUMO+1轨道,我们发现LUMO+1轨道和LUMO轨道分布存在显著差异,SNAr反应中心的2位碳原子上均有LUMO+1 lobe,而LUMO lobe基本不位于2位碳原子上。
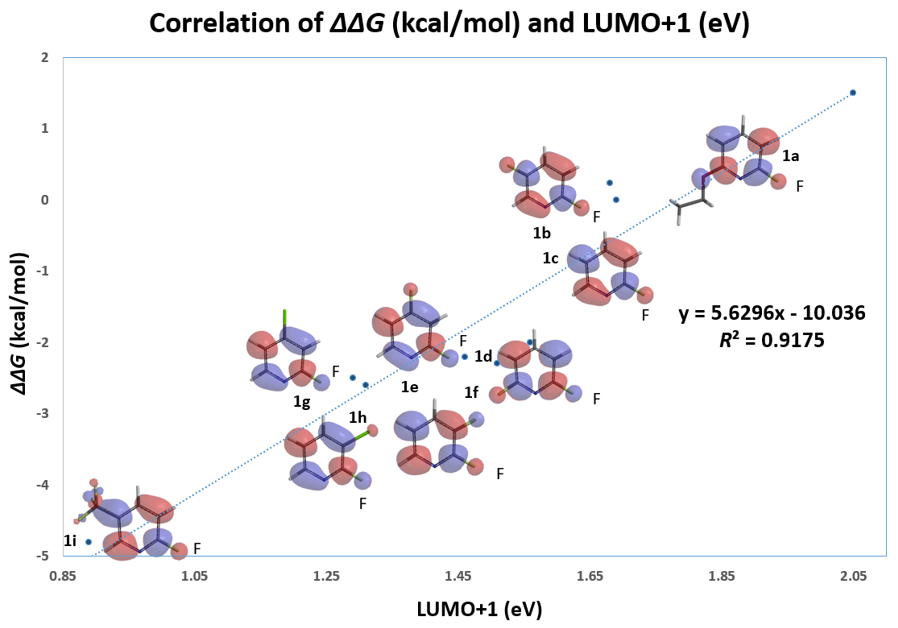
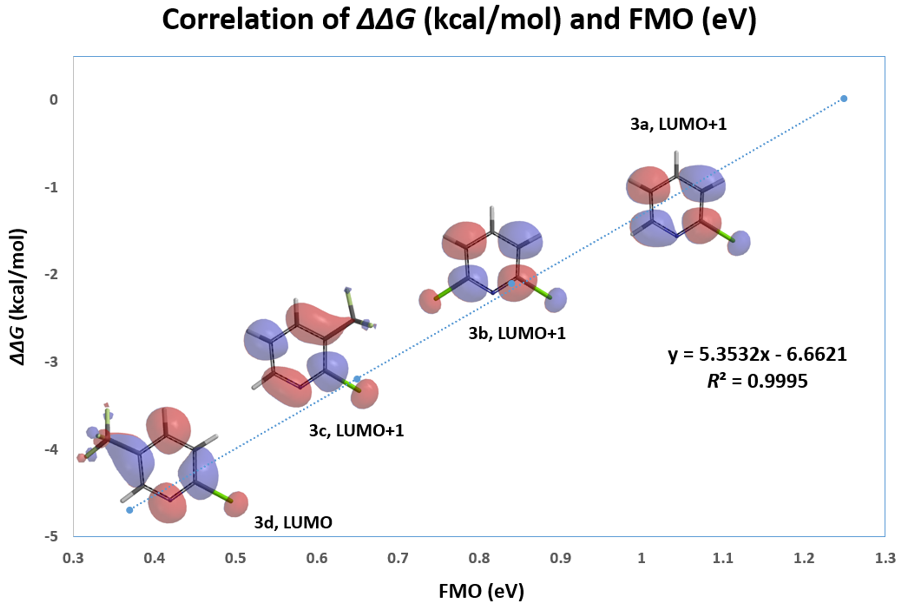
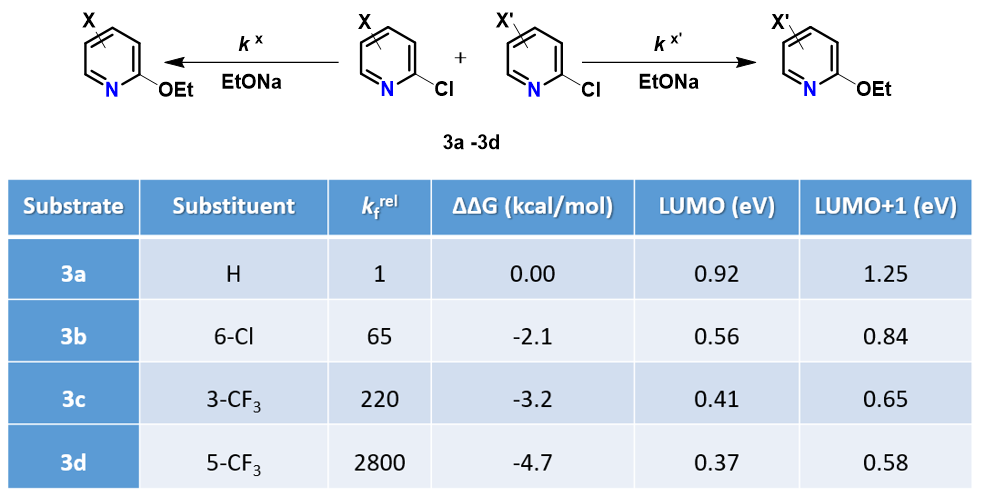
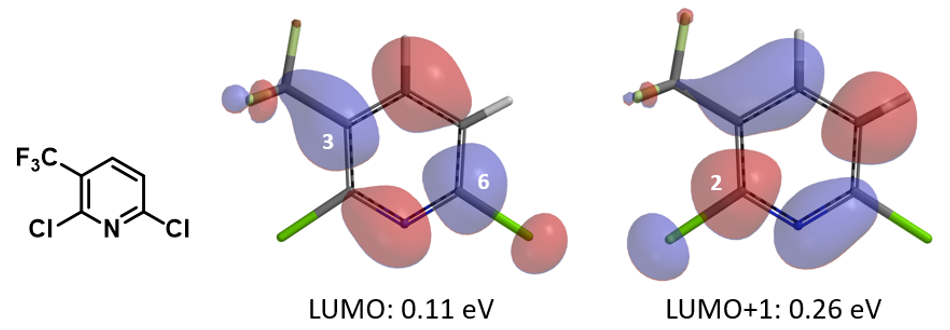
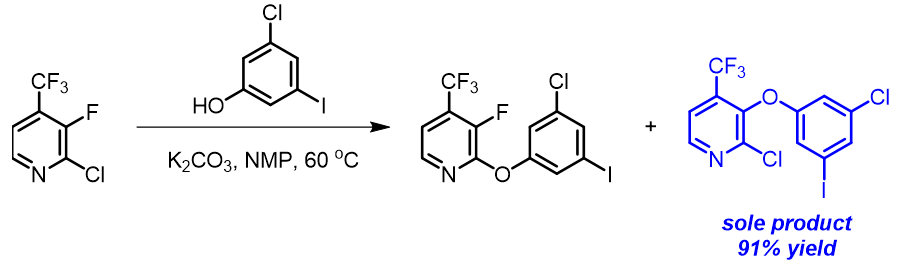
芳香亲核取代反应受底物的LUMO能级控制,我们需要通过选择合适的前线轨道,才能够看到其能级和反应的相对活化能的良好相关性。合理选择QM计算出来的参数对于用机器学习预测反应选择性的成功与否也至关重要[4]。
本文由陆颖、郑重、卫小文编撰。
参考文献:
[1] J.A. Joule & K. Mills. Heterocyclic Chemistry 5th Ed.Chichester, West Sussex, UK: Blackwell Publishing Ltd., 2010; pp 25.
[2] M. Schlosser, T. Rausis, Helv. Chim. Acta. 2005,88, 1240-1249.
[3] G. Koleva, B. Galabov, J.I. Wu, H.F. Schaefer III, P.R. Schleyer J. Am. Chem. Soc. 2009, 131, 14722-14727.
[4] (a) T. Stuyver, C.W. Coley, J. Chem. Phys. 2022,156, 084104. (b) Y. Guan, C.W. Coley, H. Wu, D. Ranasinghe, E. Heid, T.J. Struble, L. Pattanaik, W.H. Green, and K.F. Jensen, Chem. Sci. 2021, 12, 2198-2208.
[6] L.C. Campeau, Q.H. Chen, D. Gauvreau, M. Girardin, K. Belyk, P. Maligres, G.Y. Zhou, C.Z. Gu, W. Zhang, L.S. Tan, P.D. O’Shea, J. Org. Process Res. Dev. 2016, 20, 1476-1481.
