












网站维护
系统内容更新/升级中


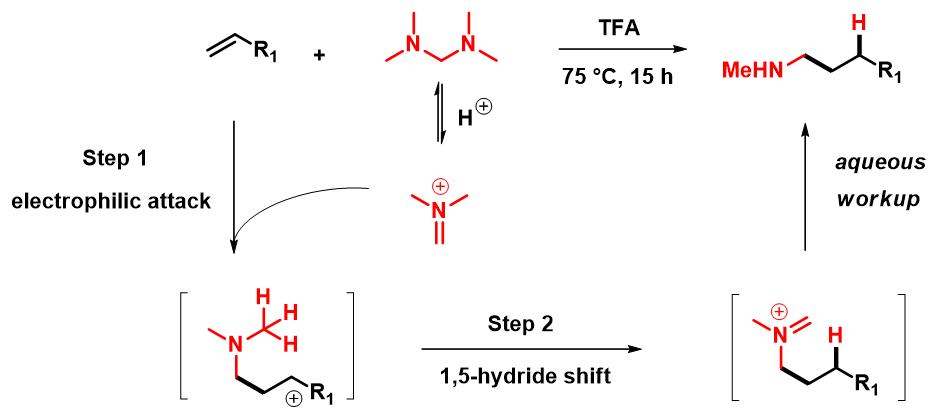
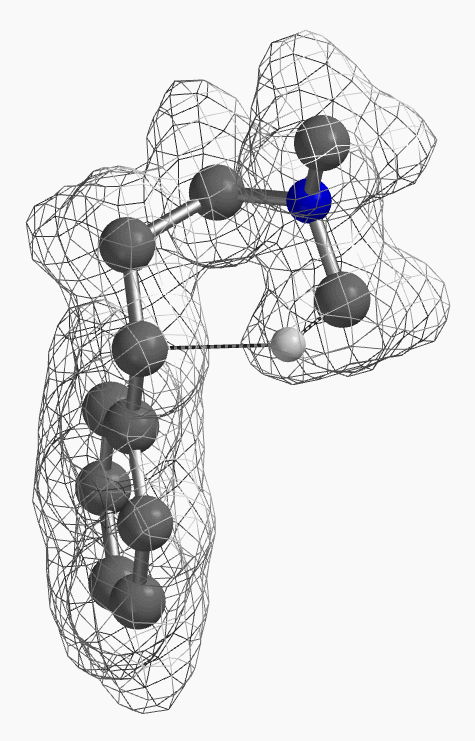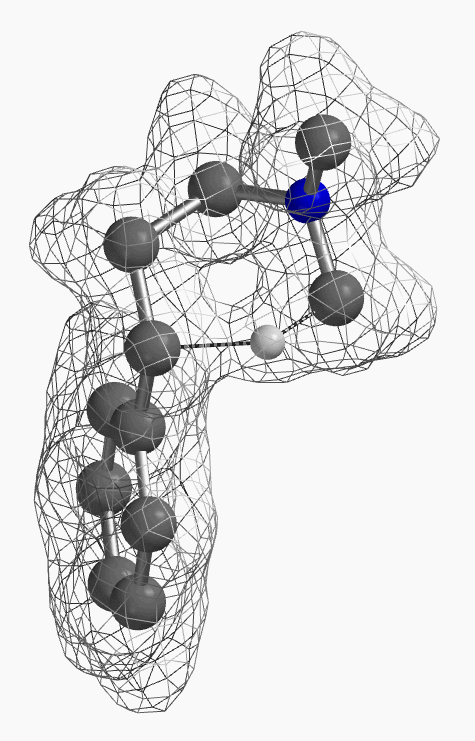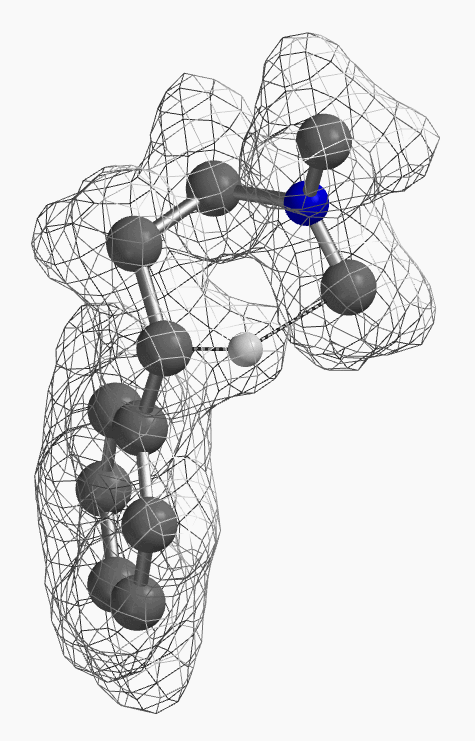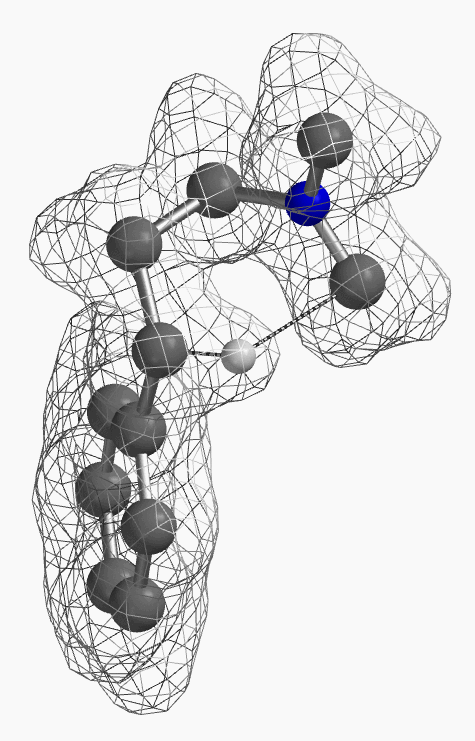
构建C(sp3)-N键的方法有很多种,除了经典的SN2反应外,含N片段对烯烃的直接加成也是常用的方法之一。金属催化的烯烃直接氢-胺化构建C(sp3)-N键已经被广泛研究[1],而烯烃的氢胺甲基化却鲜有报道。已知的报道中,大多需要用到过渡金属催化,部分文献报道光催化条件也可以促进这一转化,但需要烯烃底物有较高的活性。Nuno Maulide小组报道[2],使用Eschenmoser盐仅需酸催化,便可实现烯烃或者炔烃的氢胺甲基化。此方法原料廉价易得,底物官能团兼容性好、选择性高。与经典的SN2方法相比,消除了潜在的二甲基或季铵化的风险,适合放大生产,具有很高的实用性。文献假设的反应机理如图1所示,主要分为亲电加成和[1,5]-氢迁移两步:
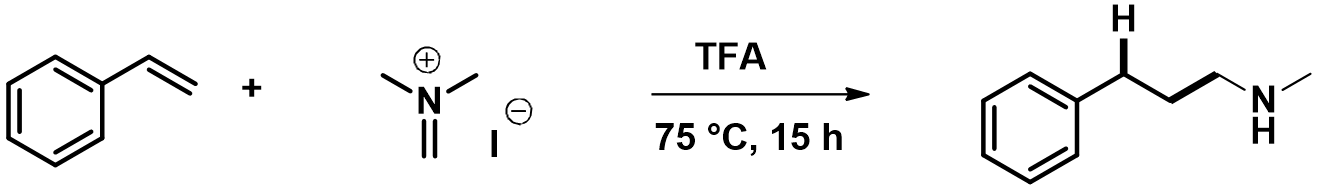
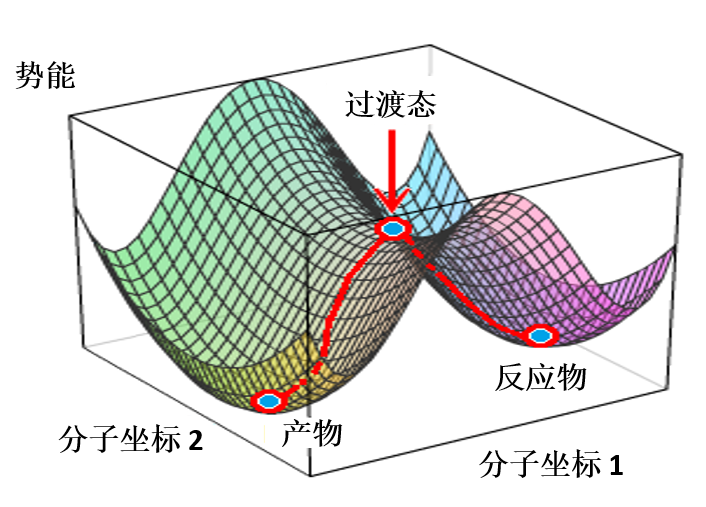
今天,我们将利用QM来验证文章中提出的机理是否合理?
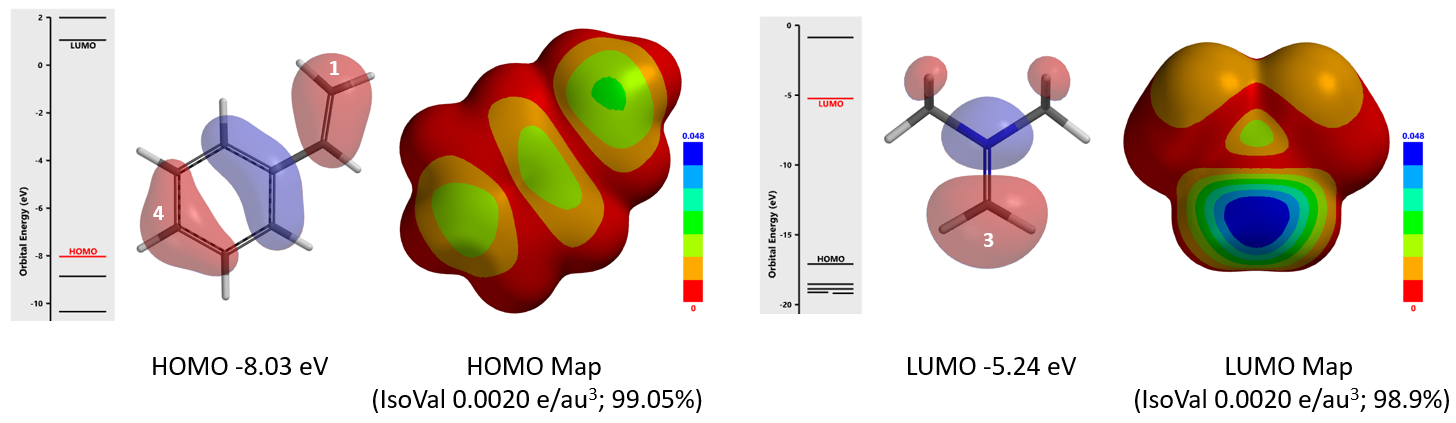
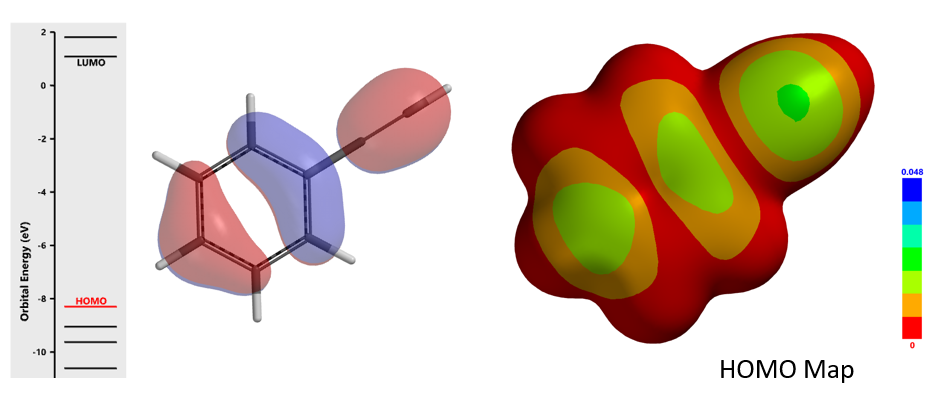
苯乙烯的HOMO(-8.03 eV)和亚胺正离子的LUMO(-5.24 eV)能量差仅为2.79 eV,易于成键[4]。苯乙烯的HOMO lobe显示电子主要分布在C-1和C-4上且大小差不多,进一步结合苯乙烯的HOMO map发现,在环外双键C-1位置的密度更高,说明C-1更容易被亲电试剂进攻。亚胺正离子的LUMO lobe主要分布在亚甲基碳原子上,说明该碳原子发生亲电反应的活性更高。以上前沿分子轨道分析和文章报道的亲电加成反应位点一致。
接下来,我们对亲电加成一步的能量变化曲线进行了计算。将起始底物苯乙烯C-1和亚胺离子C-3的距离变化设定为从彼此无相互作用的3.5 Å到共价键的成键距离1.5 Å,步长为0.1 Å。如图3所示,Step 1的活化能约为13 kcal/mol,属于该实验条件下可跨越的能垒; 接着将得到的能量峰值对应的结构B进行过渡态结构优化,并计算其红外振动, 结果表明该结构有且只有一个虚频 i299 cm-1,验证了该路径的合理性[5]。
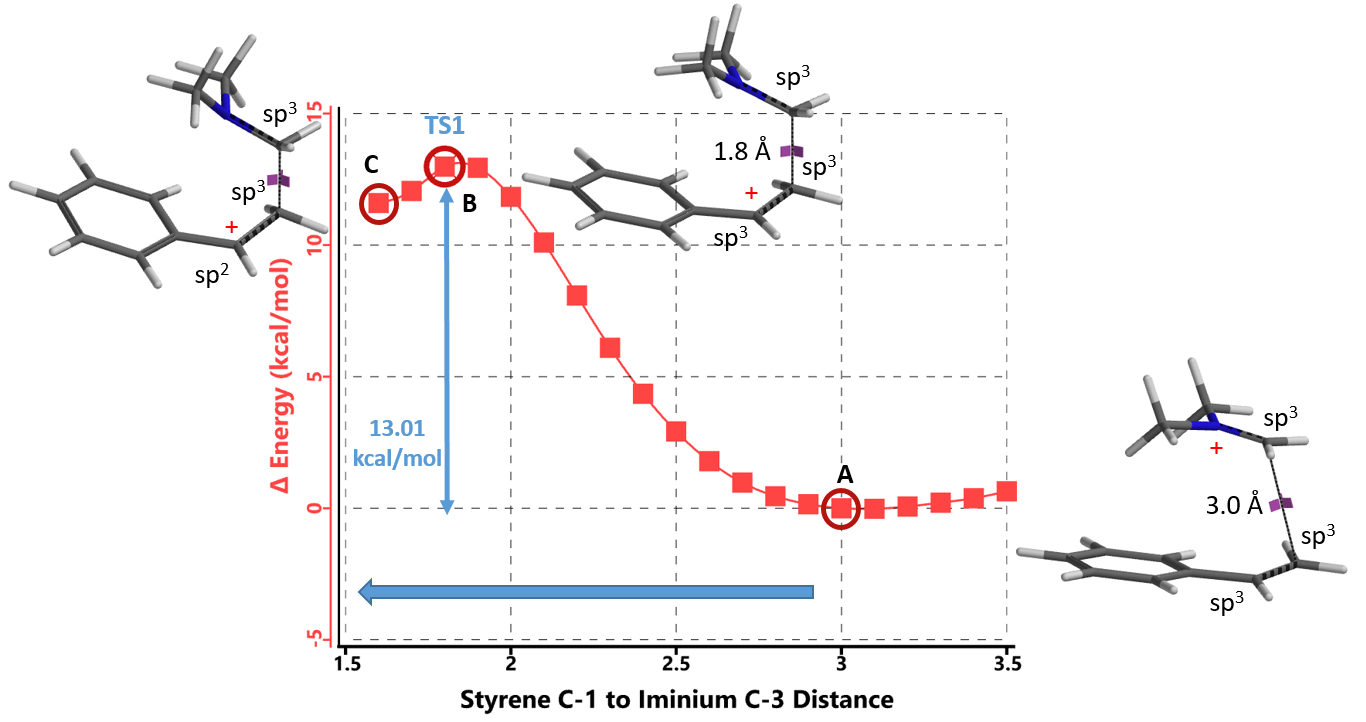
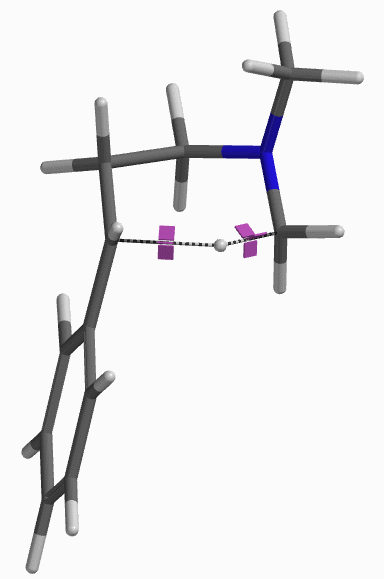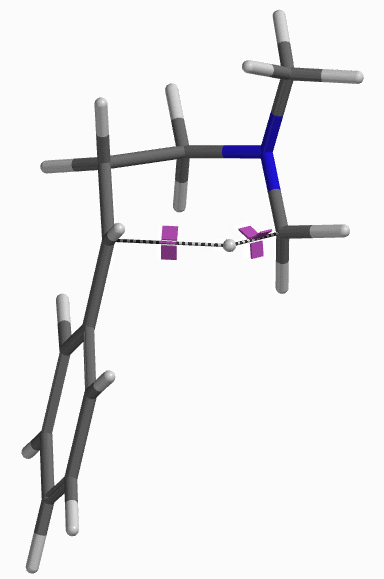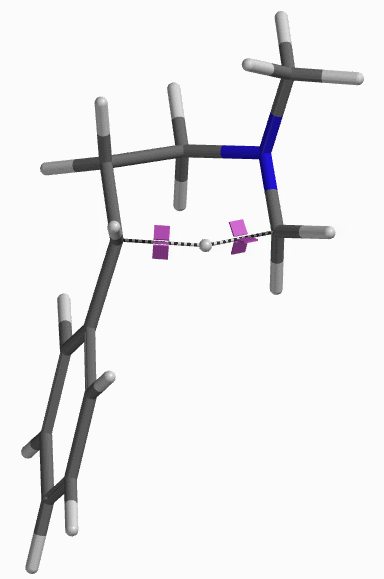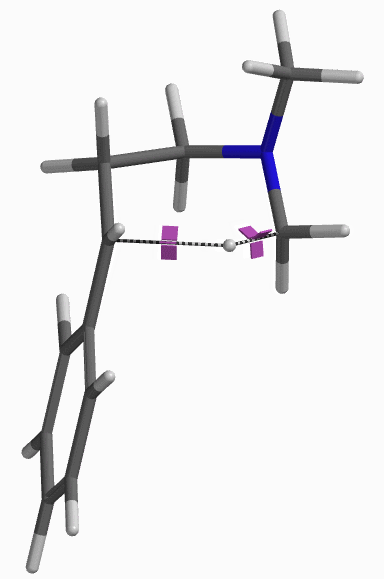
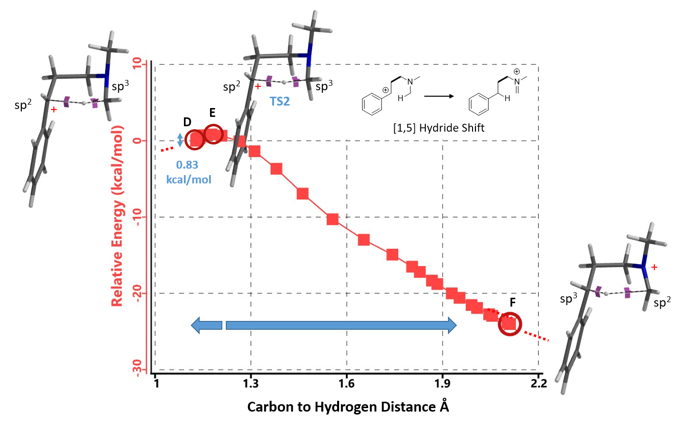
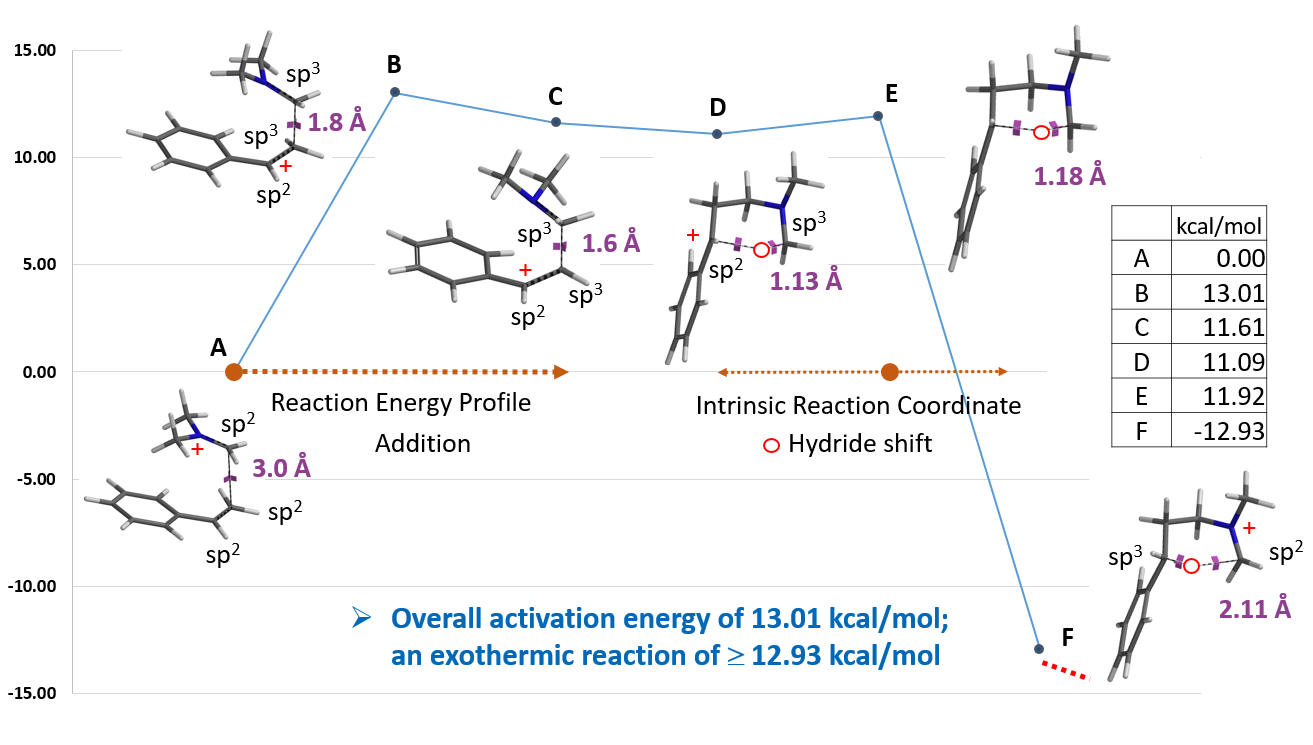
纵观整个反应历程,其总的活化能为13.01 kcal/mol,反应为放热反应,其总的相对能量变化为大于等于12.93 kcal/mol。
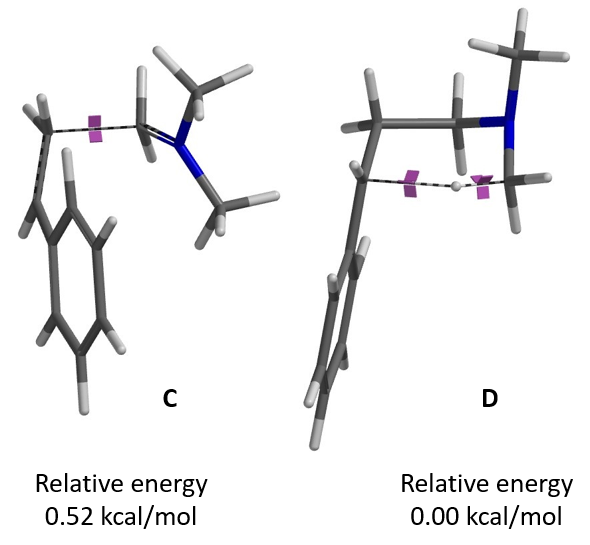
总结一下,本文通过对反应底物的HOMO Map和LUMO Map确认了亲电加成一步反应的位点,并利用活化能计算,从能量上验证了亲电加成一步机理的合理性。接着,经过计算确认和优化了[1,5]-氢迁移一步反应的过渡态结构,并由此出发计算IRC并反推出反应物和产物结构; 发现为了便于氢迁移反应的发生,亲电加成产物的构象发生了扭转的现象,氢迁移一步的反应能垒较低,反应易于发生。综合以上分析,文章中作者们所建议的机理与我们的QM计算结果相配。
参考文献及脚注:
[1] C. Michon, M.-A. Abadie, F. Medina, F. Agbossou-Niedercorn, J. Organomet. Chem., 2017, 847, 13. J.P. Huo, G.Z. He, W.L. Chen, X.H. Hu, Q.J. Deng, D.C. Chen, BMC Chemistry, 2019, 13, article 89.
[2] D. Kaiser, V. Tona, C.R. Goncalves, S. Shaaban, A. Oppedisano, N. Maulide. Angew. Chem. Int. Ed. 2019, 58, 14639.
[3] Spartan'20 Tutorial and User's Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp. 60 & 365. LUMO map 是将LUMO的绝对值映射到分子的电子密度表面上。一般我们用蓝色代表高LUMO值,用红色代表低LUMO值。LUMO所覆盖的部分意味着该区域有空位可以接纳一对电子,因此LUMO Map中蓝色富集的区域会有比较高的可能性接收亲核进攻。HOMO map 同理。
[4] L.G. Zhuo, W. Liao, Z.X. Yu, Asian J. Org. Chem. 2012, 1, 336 及 QM 魔法小课堂第二十五章评估反应活性的简单方法—LUMO和HOMO的能量差。
[5] Spartan'20 Tutorial and User's Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp. 158, 442, 459 & 536 及 QM 魔法小课堂第二十二及二十四章有关过渡态和虚频的计算。
[6] K. Fukui, Acc. Chem. Res. 1981, 14, 368. S. Maeda, Y. Harabuchi, Y. Ono, T. Taketsugu, K. Morokuma, Int. J. Quantum Chem.2015, 115, 258. 内禀反应坐标(IRC)是福井谦一于1970年提出的:它由过渡态(一阶鞍点)出发,是质量加权坐标下连接势能面相邻两个极小点的能量最低的化学反应路径(势能面上的最陡下降路径)。
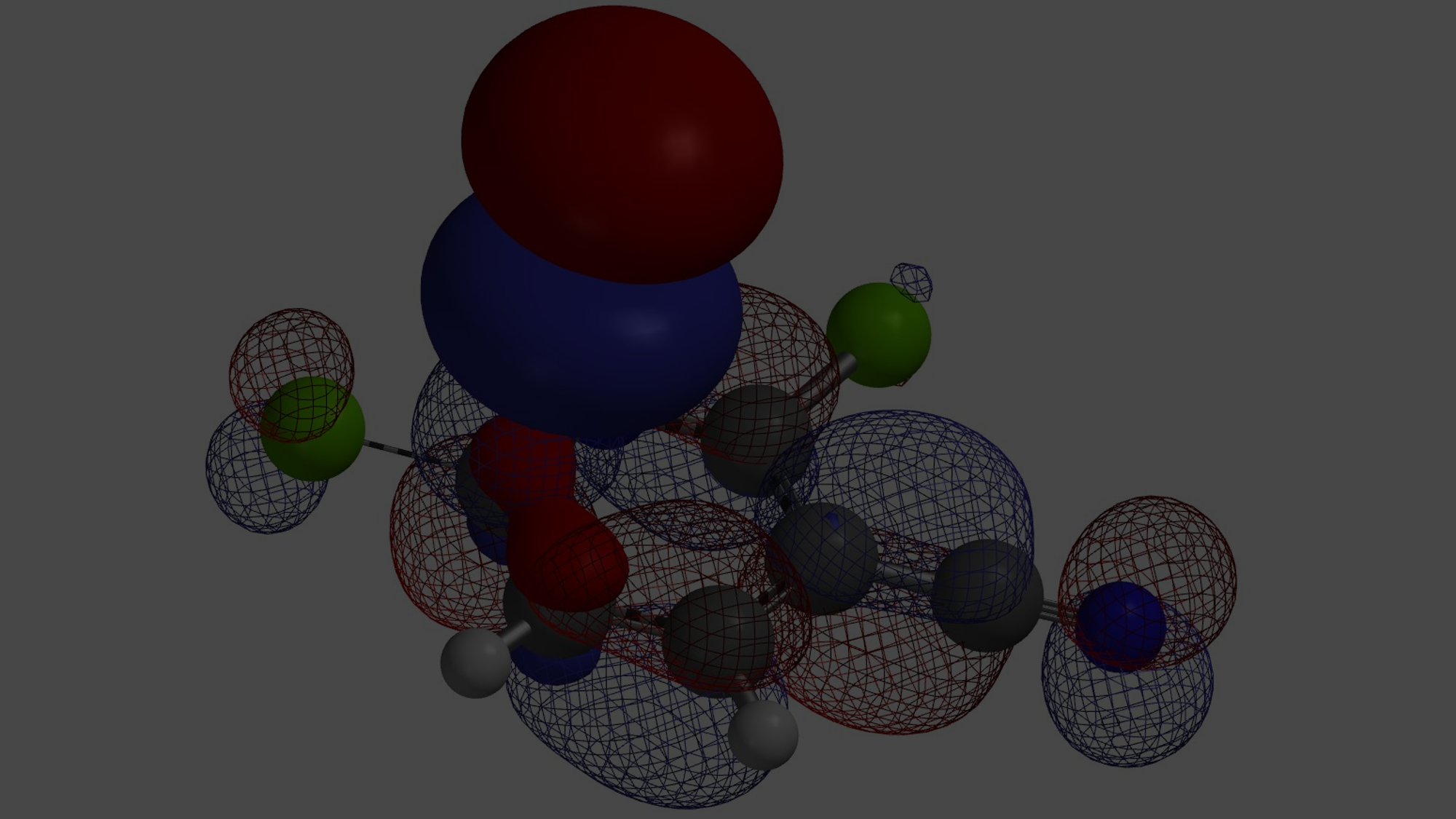
[7] 如图12所示为氢从D迁移到F过程中电子密度的变化。电子伴随着氢从氮甲基碳迁移到苄基碳上。(为了便于观察,除这一迁移氢外,分子中其它氢没有在图中显示出)。
本文由张叶霞、董立亭、赖光华、石谷沁、卫小文编撰。
