












网站维护
系统内容更新/升级中



随着科学技术的发展,药物研究领域的格局逐渐发生着变化。一种双功能小分子PROTAC介导的靶蛋白降解技术克服了传统的小分子药物研发所存在的较大局限性,因此受到业界的广泛关注。该技术利用双功能小分子PROTAC胶合靶蛋白和E3连接酶,借助细胞泛素—蛋白酶体降解系统,清理靶蛋白,进而调控疾病相关蛋白浓度水平。
作为最早从事PROTAC相关研究的科学家之一,药明康德药物研发国际服务部副总裁Dave Madge(帝国理工学院药物化学博士)表示,这项技术最吸引人的地方在于它突破了传统成药原则,使攻克“不可成药的”靶点成为了可能。从2016年起,药明康德就开始布局介导靶蛋白降解的PROTAC分子相关的技术,积累了大量的成功经验,已打造出一个较完善的集成化赋能平台。药明康德所拥有小分子药物研发平台集发现、合成、分析纯化和测试等能力于一体,并且具备DEL筛选、生物物理学、结构生物学和体外药理学等领域新技术,在化合物发现上拥有独特的优势。药明康德打造的一站式PROTAC分子药物研发平台,为介导靶蛋白降解的PROTAC技术助力,为新时代的新药研发赋能。
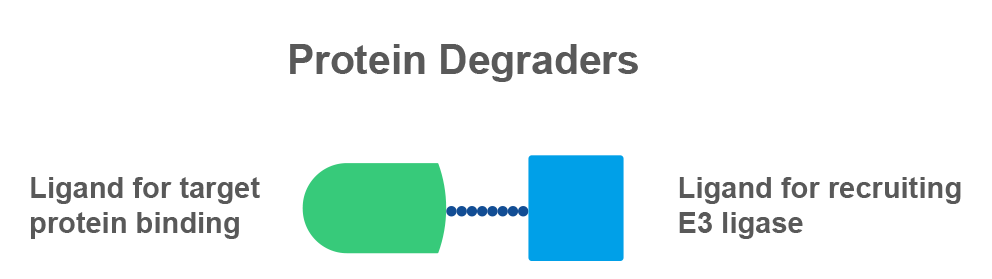
众所周知,细胞每时每刻都会生产新的蛋白,一些使用过或表达出错的“垃圾蛋白”需要及时被降解,从而维持胞内的蛋白稳态。泛素-蛋白酶体途径(Ubiquitin-proteasome pathway, UPP)是细胞发挥该功能的主要方式之一。
受“分子胶水”的启发,融合了靶蛋白识别和介导蛋白降解的PROTAC(PROteolysis TArgeting Chimera)技术应运而生。PROTAC分子像月老手中的红线,一端是结合靶蛋白的配体,另一端是结合泛素连接酶E3的配体,二者通过链接分子相连。PROTAC分子进入细胞后,一端结合靶蛋白(Protein of Interest, POI),另一端去结合泛素连接酶E3,帮助靶蛋白和E3连接酶“牵手”成功,形成POI-PROTAC-E3连接酶的三元复合物,UPP介导靶蛋白POI被贴上泛素“标签”,带有多个泛素“标签”的POI会被蛋白酶体识别并降解,从而使靶蛋白的浓度被选择性地降低。
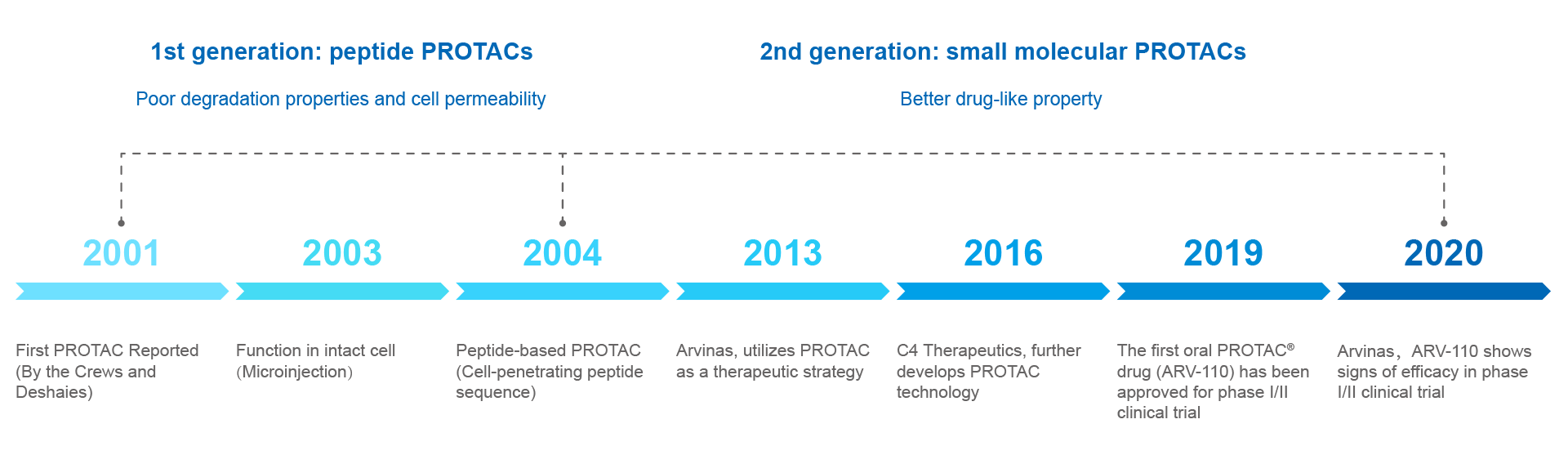
回顾PROTAC技术发展历史,一些里程碑式的事件值得铭记。
2001年是PROTAC技术的元年,Crews和Deshaies团队利用了含SCFβ-TRCP的E3诱导了MetAp-2的降解,首次引入了靶向嵌合蛋白水解(PROTAC)一词但是由于早期的PROTAC分子多为小肽,细胞渗透性和细胞活性表现不佳,无法应用到药物的开发中。
2004年是一个分水岭,之前的PROTAC分子被称为第一代PROTAC,作为proof-of-concept是成功的,但需要相对浓度比较高才能奏效,且细胞穿透力比较差。但随着技术发展,细胞穿透性很高的第二代PROTAC出现了。2013-2016年间,Arvinas公司和C4 Therapeutics公司先后成立,它们都专注于PROTAC技术,大力发展创新药物,使得PROTAC技术得以迅速地发展,为新药研发打开了新篇章。此后,PROTAC的技术不断更新。
2019-2020年,药明康德合作伙伴之一Arvinas公司研发首款口服PROTAC®药物ARV-110(拟用于治疗去势抵抗性前列腺癌)在一期临床试验中取得显著的疗效,为广大患者带来福音。
在成千上万种致病的蛋白中,传统小分子药物大多数情况下仅能作用于其中位于细胞表面的一小部分,而绝大多数致病蛋白都位于细胞内或核内,且表面相对平滑,生物大分子和小分子药物皆无能为力。而泛素化参与调控细胞周期、增殖、凋亡、分化等大量生命活动,在蛋白质的降解等过程中起重要作用,与肿瘤、心血管、自身免疫等疾病的发病密切相关。
PROTAC技术利用人体泛素—蛋白酶体降解系统,打破了药物设计的传统观念,通过技术的革新,重新定义了小分子药物,为新药研发开拓了新的方向。大量临床前和临床研究表明,PROTAC小分子药可采用口服在内的多种给药途径,给药后在人体组织器官中分布广泛,除此之外PROTAC小分子药还具有以下几点优势[1]:
1. 用量小,利用率高。
2. 克服以往难以成药的靶点。
3. 避免靶蛋白突变产生的耐药性。
但机遇总是伴随着挑战,作为一种新兴的药物研发技术,PROTAC目前遇到以下挑战:1. E3连接酶种类过于繁多。
E3连接酶的种类繁多,超过600种。目前,指导不同E3连接酶与靶蛋白的配对组合仍基于传统经验,效率低,不利于E3连接酶配体的开发和利用。
2. 配体小分子不易寻找。
3. 降解水平的不确定性。
4. 耐药性的产生和克服方法;
从2016年起,药明康德就开始布局介导蛋白降解的PROTAC分子相关的技术,积累了大量的成功经验,已打造出一个较完善的整合化赋能平台。作为药明康德布局PROTAC分子相关研究的科学家之一,药明康德药物研发国际服务部高级主任周磊博士表示,基于模块化的工作流程,在药明康德不同部门的分工和配合下,首批PROTAC候选药物分子仅用了三四周左右的时间,便交到了合作伙伴手上。而随着对PROTAC了解的逐渐深入,药明康德PROTAC平台的效率也在稳步提升。
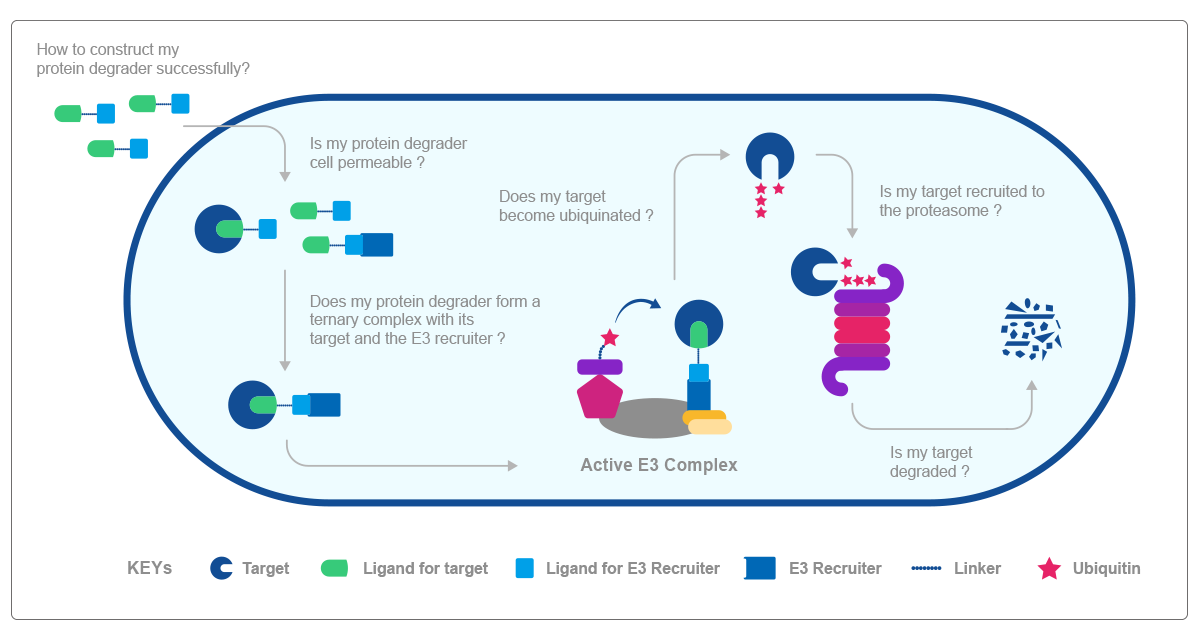
上图为PROTAC分子介导靶蛋白降解技术的所含关键步骤,归纳如下:
第一步:如何成功构建PROTAC分子
第二步:PROTAC分子能否成功穿透并进入细胞;
第三步:一旦进入细胞,靶蛋白-PROTAC分子- E3连接酶的三元复合物能否形成;
第四步:能否形成活化的E3复合体;
第五步:靶蛋白能否被泛素化;
第六步:泛素化的靶蛋白能否被蛋白酶体识别并结合;
第七步:靶蛋白是否发生降解。
与传统药物的药理学相比,PROTAC分子对配体本身的功能活性要求不高。因此,通过直接检测靶蛋白与配体之间的相互作用,就可以判断PROTAC分子是否构建成功。目前,药明康德已经研发出一套适用于蛋白质等生物大分子的PROTAC分子构建体系,包括靶蛋白的筛选、靶蛋白的制备和连接子的选择等,同时兼顾靶蛋白-PROTAC分子-E3连接酶的三元复合体的形成。
如上图所示,靶蛋白确定后,针对靶蛋白配体的选择、E3连接酶配体的选择和优化等,药明康德提供了DNA编码化合物库(DNA Encoded Library, DEL)、基于片段的筛选(Fragment-based drug design, FBDD)、基于结构的筛选(Structure-based drug design, SBDD)和虚拟筛选(Computer-aided drug design, CADD)等方法。
1)DEL筛选
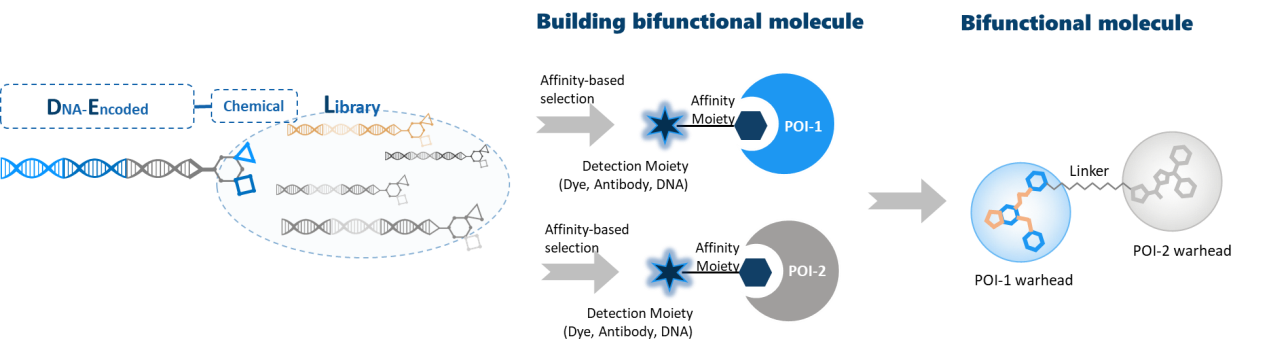
DEL是一种用来筛选小分子药苗头化合物的新技术。药明康德特色的DEL库,基于大量研究和文献,在100多个成药性良好的母核结构、6 000多个独有的官能团和35 000多个精选商售的分子砌块基础上,应用 “Split & Pool”原理,化合物容量超过900亿个,覆盖了绝大多数成药化合物的化学空间。不仅如此,所有的化合物都连有一条独特的DNA“条形码”。通过解码这段DNA序列,就能快速找到与靶蛋白结合的小分子。
DEL筛选打破了以往一对一的筛选模式,在以下几个方面的做到了创新:
基于亲和性筛选,确保靶蛋白和配体间存在结合力
数量庞大的化合物库实现了小分子结构的多样性和创新性
现成的连接体简化了双功能小分子的设计和构建
筛选所需蛋白耗量少(5ug/条件),周期短(4周)和多靶点/条件平行筛选
除此之外,药明康德HitS事业部可以根据客户的需求,提供DEL库的定制服务,还提供针对筛选结果的“off-DNA”检测(另外合成不含DNA标签的化合物小分子),借助生物物理学和体外试验确保筛选结果的准确性。
2)FBDD筛选
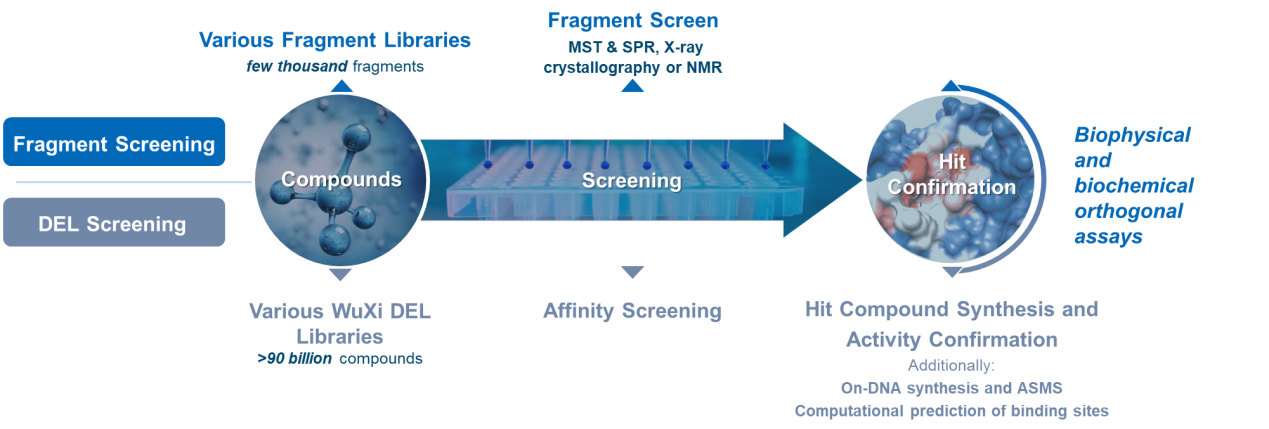
FBDD筛选适用于研究蛋白相互作用(PPI),是小分子化合物发现的主流方法之一。它利用核磁共振技术(NMR)、表面准粒子共振技术(SPR)、X-射线单晶衍射(X-ray)等方法筛选出与靶蛋白相互作用的小分子片段,再基于小分子的结构信息对活性片段进行优化,进而得到更高活性的小分子化合物用于新药的研发。
一方面,由于FBDD小分子的分子量低(通常<250 Dalton),结构简单,只需要较短的时间就能够发现蛋白质表面独特的亚活性结合位点;另一方面,筛选到的小分子化合物质量高,便于药物后续的结构改造与优化。
药明康德FBDD库中约有三千多个小分子,这些精心挑选的小分子结构典型,成药性好,便于优化。药明康德DEL实验室的负责人Nuska Tschamer建议,FBDD筛选需搭配最先进的生物物理学方法,只有灵敏度足够高,才能可以检测到小分子和靶蛋白之间的特异性结合。
3)SBDD筛选
SBDD筛选是从靶蛋白和配体的共晶三维结构出发,以结构生物学为基础的药物设计。靶蛋白的三维结构,通常可以通过X-射线晶体学(X-Ray crystallography)、核磁共振(NMR)或同源蛋白质结构预测等方法得到。而后可以通过实验测定或用分子建模计算出候选靶蛋白与配体间的相互作用,在此基础上进行理性药物设计。SBDD筛选不仅依赖于靶蛋白结构信息,还得助于靶蛋白-配体间的相互作用的热力学和动力学信息。目前,主流技术仍采用X-射线晶体学进行结构解析。
4)CADD筛选
CADD筛选是一种以计算机化学为基础,与SBDD相结合的虚拟筛选方法。这种筛选策略利用计算机上的分子对接软件模拟靶蛋白与候选药物之间的相互作用,计算两者之间的亲和力大小,通过设定标准值,降低试验筛选的化合物数目,提高先导化合物发现效率。
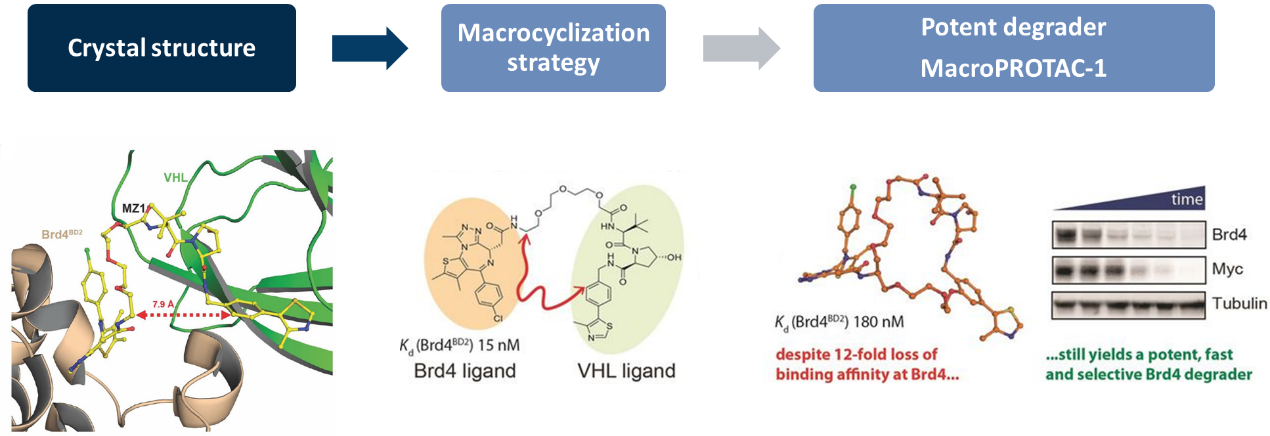
例如,已知了MZ1(一种连接Brd4BD2和VHL的PROTAC分子)、Brd4BD2和VHL三元复合物的晶体结构。William Farnaby(邓迪大学生命科学学院的,化学生物学和药物化学学博士)和其团队经过设计和分析,将双功能小分子MZ1的结构进行环化处理,从而达到优化的效果。该设计后续被验证获得成功,他们获得了名为MacroPROTAC-1的高效蛋白降解剂。尽管亲和力受到影响,但它在降解Brd4蛋白时,效率更高、特异性更好[2]。
5)计算机模拟三元复合体的形成
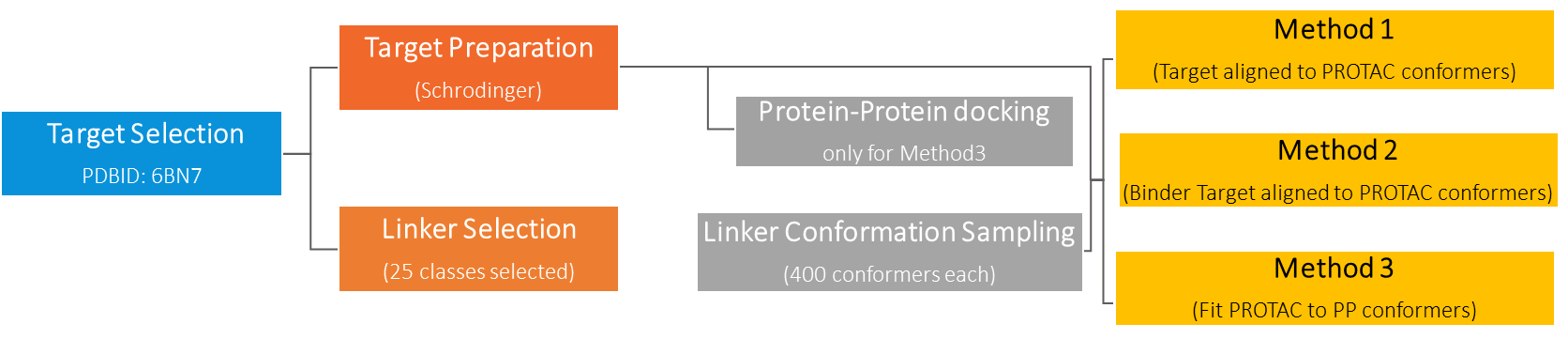
目前有三种主流方法可以模拟基于PROTAC分子的三元复合体的形成路径[3]:
方法1:先构建靶蛋白配体—连接子—E3配体的PROTAC分子,再将构建好的PROTAC分子的两端分别结合靶蛋白和E3连接酶;
方法2:先构建靶蛋白与其配体和连接子的复合体,再用构建好的复合体连接对应的E3连接酶及其配体;
方法3:先分别构建两个复合体,即靶蛋白及其配体复合体以及E3连接酶及其配体复合体。由于靶蛋白和E3连接酶之间可能存在对接作用,靶蛋白配体和E3连接酶配体之间距离也很近,此时用连接子链接两个复合体中的配体,形成靶蛋白配体-连接子-E3配体的PROTAC分子。
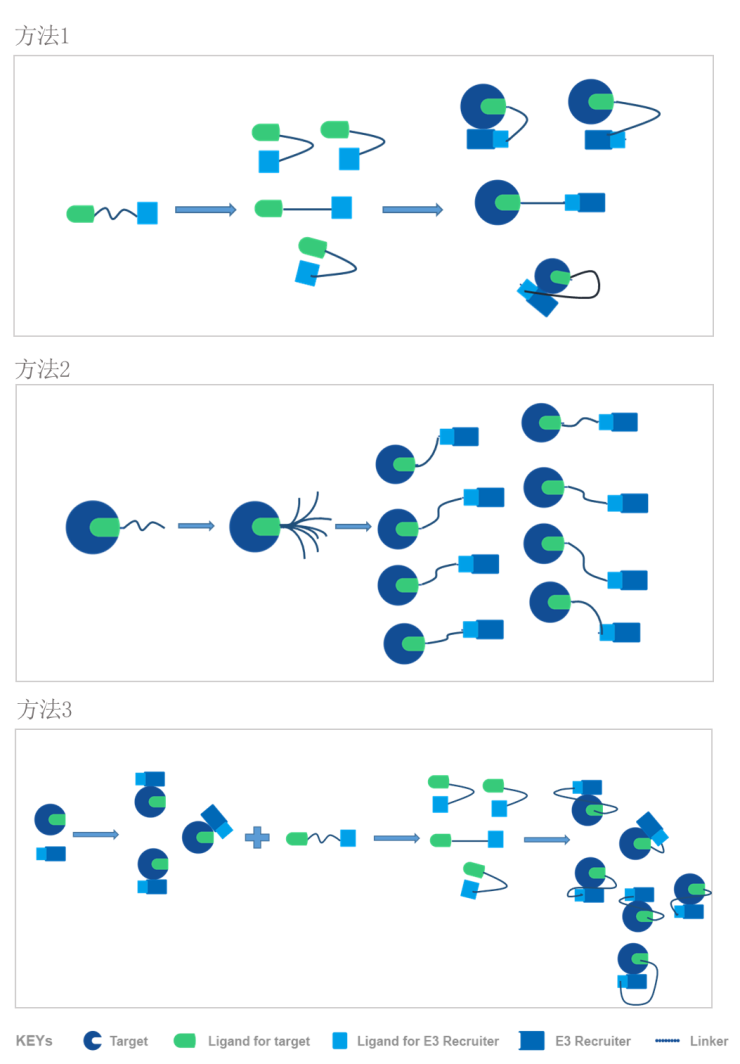
上述三个方法也并非完全独立,比如上述方法1也可以被运用在方法3中。William团队成功验证了方法3的有效性,并表明基于蛋白质对接原理的方法3,精确性更好,可作为目前主流构建方法的补充方法,用来检验假设的合理性以及优化双功能小分子的设计原则。
药明康德化学服务部(CSU)还为构建PROTAC分子配备了一个完整的工具分子库,可供直接使用。包括成熟的E3连接酶配体如VHL、CRBN、MDM2、IAP等,50多种常用的连接子如PEG、烷基、刚性连接体等,以及100多个基于文献报道的前体分子(连接体+E3配体)。此外,化学服务部在PROTAC分子的订制合成方面也有非常丰富的经验,可以提供各种个性化以及从毫克到公斤级的定制服务。目前,药明康德正努力扩大生产规模,提升符合国际GMP标准的生产能力。
除此之外,药明康德辉源生物(HDB)也为PROTAC分子的研发提供技术支持,如利用AlphaScreen、TR-FRET、荧光偏振(FP)、放射性分析等检测靶蛋白-PROTAC分子-E3连接酶三元复合体的形成,以及利用NanoBRET技术检测胞内的各项指标等。
针对第二步,生物发光共振能量转移技术(NanoBRET)可以作为一个有力的工具来评估PROTAC分子的细胞通透性。活细胞中,由融合到VHL的NanoLuc®荧光素酶作为能量供体,荧光细胞渗透性小分子(VHL示踪剂)作为能量受体。细胞渗透性较强的PROTAC分子将与胞内VHL示踪剂竞争性结合VHL,导致BRET信号强度的减弱,从而可以判断双功能小分子的膜透性。
靶蛋白-PROTAC分子-E3连接酶三元复合体经历以下过程:
靶蛋白与PROTAC分子形成二元复合物,再与E3连接酶结合形成三元复合体;同时, E3连接酶也可以先与双功能小分子形成二元复合物,再与靶蛋白竞争,形成三元复合体。
![]()
上述过程中的所有反应伴随能量的交换,测定不同反应中的动力学参数——Kd值,再引入协同性参数α(cooperativity α),就可以表征二元复合体和三元复合体的形成情况。常用的测定反应动力学参数的方法包括如表面准粒子共振(SPR)、微量热泳动(MST)和等热滴定(ITC)。
AlphaLISA是一种基于微珠的化学发光新型检测技术,相较传统的AlphaScreen,灵敏度更高,准确性更好。比如在测定VHL- MZ1-Brd4三元复合体的形成时,AlphaLISA将VHL E3连接酶作为供体,将Brd4蛋白的微珠作为受体。如果VHL E3和Brd4之间存在相互作用,供体和受体微珠将靠近,在615nm或者680nm光照下,激发级联放大的化学反应,产生大量信号扩增,直观反映为吸光度值显著增加。
HiBiT 和LgBiT是鼎鼎大名的NanoLuc® 萤光素酶的两个亚基,前者仅含 11 个氨基酸的标签,是一个理想的分子标签。同时,单独的亚基不具有发光活性,只有二者组合后才能发挥萤光素酶的活性,并且这两个亚基之间具有极高的亲和力。
药明康德HDB打造的基于HiBiT 和LgBiT之间亲和力的 “二合一”系统,利用构建好的 Knock-in 细胞系或者含 HiBiT 蛋白标签载体,标记靶蛋白,通过检测特异性启动子开启或者关闭(控制LgBiT表达)情况下系统内的荧光水平,实时监测胞内靶蛋白的降解情况。更为重要的是,该方法支持在同一细胞系中进行筛选和蛋白质降解动力学研究,易于后期的数据整理和比较。
未来,经典的泛素-蛋白酶体降解体系仍将是该领域的热点。因此,如何扩展E3连接酶配体的功能就显得非常重要。例如,HECT结构域酶可与泛素分子相互作用,进而形成硫酯键;环形结构域能催化泛素的直接转移;CDC20是所有癌细胞的必需组分,在大多数正常组织中表达量较低。这提示我们,能否开发与癌细胞生存至关重要的E3连接酶,避免耐药性的产生[4]。
溶酶体是另一种细胞降解胞内外物质的主要途径。目前,已有报道介绍了几种基于溶酶体降解蛋白的新模式。例如,溶酶体靶向嵌合体(Lysosome targeting chimera,LYTAC)利用聚糖标签来标记靶蛋白,利用受体介导胞内溶酶体降解靶蛋白;自噬靶向嵌合体(Autophagy-targeting chimera,AUTAC)会与靶蛋白结合并用一个类似S-鸟苷酸化的降解标签标记靶蛋白,这种翻译后修饰可触发靶蛋白的K63多泛素化,进而引发降解;自噬体绑定化合物(Autophagosome-tethering compound,ATTEC)会与靶蛋白和LC3相互作用,通过将靶蛋白与吞噬体或自噬体绑定,使蛋白降解[5]。
为了实现“每一种药物都可以制造,每种疾病都可以治疗”的愿景,通过赋能全球制药、生物科技和医疗器械公司,药明康德致力于推动新药研发进程,为患者带来突破性的治疗方案。药明康德相信,双功能小分子这一富有想象力的新模式将赋能蛋白降解途径,为药物研发提供一种新思路。为此,药明康德借助最先进的生物物理学、结构生物学和体外药理学方法,倾力打造了一站式的介导靶蛋白降解的PROTAC小分子药物研发平台。结合化学服务部(CSU)提供的工具分子库和定制合成服务,可以赋能合作伙伴更快更高效的推进新药研发进程,辉源生物(HDB)利用AlphaScreen、NanoBRET、NanoBiT等先进的生物物理和生物化学方法,为胞内各项指标的检测及体内药理学的研究提供技术支持。尤其是全新HitS事业部的成立,将进一步强化DEL等技术在化合物发现上的优势作用,进而帮助合作伙伴找到更多具有良好药效的介导靶蛋白降解的PROTAC小分子药物。
